
Газета «Новости медицины и фармации» Гастроэнтерология (337) 2010 (тематический номер)
Вернуться к номеру
Хронический гастрит: современный взгляд на проблему
Хронический гастрит (ХГ) и гастродуоденит — самые распространенные гастроэнтерологические заболевания. Не секрет, что зачастую врачами и пациентами ХГ рассматривается как банальное заболевание, не требующее детальной диагностики и адекватного лечения. Вместе с тем ХГ является предиктором и фоновым состоянием для язвенной болезни и рака желудка (РЖ) [1, 4, 16]. За последние годы раскрыты многие неизвестные ранее стороны патогенеза ХГ, позволяющие принципиально изменить взгляды на возможность лечения этого заболевания и осуществления профилактики рака желудка [12].
Прежде всего следует учитывать, что ХГ — клинико-морфологический диагноз, который устанавливается на основании характерной симптоматики с обязательной морфологической верификацией, именно поэтому современное определение ХГ включает преимущественно морфологические характеристики. ХГ — это хронический воспалительный процесс слизистой оболочки желудка (СОЖ), характеризующийся нарушением ее физиологической регенерации, уменьшением количества железистых клеток, при прогрессировании — атрофией железистого эпителия с развитием кишечной метаплазии, а в последующем — дисплазии [6, 10].
Хронический гастрит и H.pylori
Хеликобактериоз является так называемой медленной инфекцией, что обусловлено длительным, практически пожизненным, персистированием возбудителя в организме человека, его способностью взаимодействовать с иммунной системой и адаптироваться к ее изменениям [2, 13]. Повреждающие механизмы H.pylori-инфекции принято подразделять на прямые повреждения продуктами патогена (уреаза, вакуолизирующий цитотоксин и др.) и деструкции, опосредованные иммунопатогенетическими механизмами (антигенная мимикрия, сдвиги в запуске цитокинового каскада по провоспалительному пути и др.). Первым этапом развития инфекции является колонизация слизистой оболочки бактериями, для чего им требуется преодолеть кислотный, а затем и слизисто-бикарбонатный барьер желудка [6].
Важнейшим фактором вирулентности Н.pylori является бактериальная уреаза (мочевинная амидогидролаза). Свыше 5 % всех клеточных белков Н.pylori приходится на долю уреазы, что свидетельствует о «чрезвычайном» уровне продукции данного энзима [28]. Уреаза хеликобактерий, в отличие от других бактериальных уреаз, не только находится в цитоплазме, но и ферментативно активна на поверхности микробов. В физиологических условиях постоянно поступающая путем транссудации из плазмы крови мочевина желудочного содержимого гидролитически расщепляется уреазой Н.pylori до аммиака и угольной кислоты с последующим образованием гидроксида аммония и НСО3-аниона. Гидролиз мочевины завершается образованием щелочных продуктов, что приводит к локальному повышению рН и формированию защитного аммиачного облака вокруг микроорганизма, нейтрализующего HCL желудочного содержимого. Существует особый механизм, регулирующий гидролиз мочевины в зависимости от окружающей клетку рН по принципу отрицательной обратной связи. Уреаза, гидролизуя мочевину в микроокружении, нейтрализует пенетрацию Н+-ионов сквозь клеточную стенку бактерии, поддерживая внутриклеточный потенциал рН на уровне, необходимом для жизнедеятельности бактерий [29]. Таким образом, обеспечиваются выживание и процесс размножения микроорганизма.
Несмотря на многообразие клинических форм хеликобактериоза, во всех случаях имеет место общий механизм — воспаление СОЖ, развивающееся вслед за адгезией микроорганизма на желудочном эпителии. Морфологически такое воспаление характеризуется инфильтрацией собственной пластинки слизистой нейтрофильными лейкоцитами, лимфоцитами, макрофагами, плазматическими клетками, формированием лимфоидных фолликулов и повреждением эпителия различной степени выраженности. Нейтрофильная инфильтрация эпителия и собственной пластинки СО индуцируется посредством реализации двух различных механизмов — непосредственно Н.pylori (за счет выделения растворимого белка, активирующего нейтрофилы) и опосредованно через экспрессию эпителиоцитами интерлейкина-8 (IL-8) с последующим запуском всего провоспалительного каскада. Белок, активирующий нейтрофилы, имеется у всех известных штаммов Н.pylori, что объясняет наличие нейтрофильной инфильтрации у 100 % инфицированных. При этом вариабельность степени воспалительной инфильтрации, по всей видимости, вызвана различиями в степени адгезии и обсемененности Н.pylori, где на первый план может выступать IL-8-обусловленный механизм. Феномен хемотаксиса нейтрофилов после активации IL-8-цитокинового сигнального каскада более выражен у CagA-, VacA-позитивных штаммов. Одним из эффектов нейтрофильной инфильтрации является увеличение проницаемости эпителия для антигенов Н.pylori, вызывающих миграцию в собственную пластинку лимфоцитов, плазматических клеток и макрофагов [5, 17]. Важная особенность состоит в отсутствии спонтанного уничтожения Н.pylori под воздействием антихеликобактерных антител, что, вероятно, объясняется «недоступностью» бактерии для антител в слое желудочной слизи, невозможностью выделения IgG в просвет желудка при относительном дефиците секреторных IgA, а также антигенной мимикрией Н.pylori [37, 40]. Таким образом, неэффективный гуморальный иммунный ответ на Н.pylori и его антигены вместо элиминации возбудителя становится одним из факторов патогенеза, формируя различные клинические варианты хеликобактериоза.
Сегодня широко обсуждается вопрос о взаимосвязи CagA-, VacA-позитивных штаммов Н.pylori с язвенной болезнью двенадцатиперстной кишки, раком желудка и повышенным воспалительным ответом СОЖ (по сравнению с нетоксигенными штаммами). Имеющиеся данные противоречивы, в одних исследованиях эта взаимосвязь подтверждается, в других — напротив, опровергается, в третьих — доказательная база недостаточна для определенного утверждения. Вместе с тем большинство авторов свидетельствует о наличии ассоциации CagA (+) штаммов Н.pylori с некардиальным раком желудка [31, 39].
Каскад Р. Соrrеа и предраковые состояния желудка
Ключевым моментом в диагностике ХГ является раннее выявление предраковой перестройки желудочного эпителия. Желудочный канцерогенез — сложный и многоступенчатый процесс. Основываясь на результатах исследований, проведенных во всем мире, Р. Соrrеа в 1988 году постулировал последовательность патологических изменений в СОЖ от нормального состояния до возникновения рака (рис. 1) [19]. Согласно этой модели, ХГ прогрессирует с развитием атрофии и кишечной метаплазии (КМ). У некоторых лиц метаплазированный эпителий подвергается дальнейшим геномным и фенотипическим изменениям с развитием дисплазии и может прогрессировать в инвазивную опухоль. Впоследствии эта парадигма была модифицирована автором с включением H.pylori в начало указанной последовательности патологических изменений [20] (рис. 1).
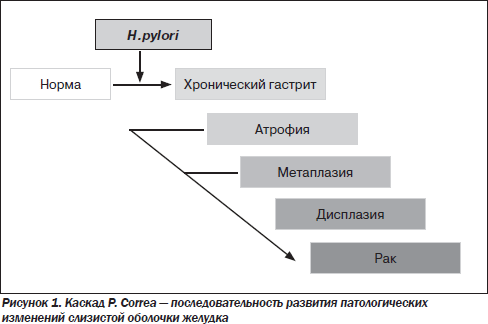
Этапы «каскада Соrrеа» — атрофия, КМ и дисплазия — сегодня рассматриваются как предраковые изменения СОЖ, т.е. морфологические изменения с высоким риском трансформации в рак желудка (РЖ) [9].
Природа атрофии
Атрофия — это утрата желудочных желез с замещением их метаплазированным эпителием или фиброзной тканью. Подобное состояние является следствием нарушенного клеточного обновления и развивается в результате изъязвления с деструкцией СО или в процессе длительного воспалительного процесса, при котором многочисленные железы разрушаются независимо друг от друга. Существуют два принципиально различных пути развития атрофии: первый, при котором железистый аппарат подвергается деструкции вследствие прямого повреждения или в результате воспалительного ответа, и второй путь, при котором избирательная деструкция специализированных эпителиальных клеток происходит в условиях сохранения стволовых клеток. Оба пути могут иметь место при хронической H.pylori-инфекции [23]. С одной стороны, бактериальные протеазы, высвобождаемые активированными нейтрофилами, могут разрушать железистый эпителий и стволовые клетки, а с другой стороны, продуцируемые аутоантитела реагируют с эпитопами протонной помпы париетальных клеток [14]. Прямое повреждение эпителия бактериальными продуктами не объясняет развития атрофии желез, так как колонизация ограничена поверхностным и ямочным эпителием [44]. Уменьшение объема железистой ткани сопровождается ее замещением фиброзной тканью. В то же время атрофию можно рассматривать как потерю функционально активных специализированных клеток, т.е. утрату париетальных и главных клеток без деструкции желез. При этом специализированные клеточные элементы фундальных желез замещаются тубулярными железистыми структурами из шеечных мукоцитов. Подобная частичная атрофия, или «преатрофия», описана у людей при аутоиммунном гастрите, а у животных часто отмечается как при аутоиммунном гастрите, так и при инфицировании бактериями рода Helicobacter. В последних случаях специализированные кислотопродуцирующие клетки замещаются слизистыми клетками («слизистая метаплазия»). Вероятным объяснением этого феномена может служить миграция пролиферирующих шеечных мукоцитов по направлению вниз к основаниям желез, но пока не ясно, является ли это начальным этапом в развитии так называемой пилоризации желез тела желудка [8, 45].
Частота развития и степень тяжести атрофии у пациентов с ХГ неуклонно увеличиваются с возрастом. При этом наблюдается распространение СОЖ антрального типа на территорию тела желудка со смещением в проксимальном направлении границы тело/антрум. В другом варианте подобная пилоризация желез тела желудка происходит мультифокально по всей СОЖ. Такая атрофия обычно максимально выражена на малой кривизне в области инцизуры. Атрофия не является результатом старения per se, поскольку у пожилых пациентов при отсутствии гастрита наблюдается нормальная кислотная продукция [24].
Главным пусковым механизмом развития атрофического гастрита считается H.pylori-инфекция, однако атрофия может также быть результатом аутоиммунного гастрита и длительного рефлюкс-гастрита. В зависимости от этиологии гастрита механизмы развития атрофии являются различными.
Потеря желез при аутоиммунном (тип А) гастрите опосредована антителами, хотя растет и число доказательств роли Т-клеточной цитотоксичности. При H.pylori-ассоциированном по-вреждении СОЖ описан феномен молекулярной мимикрии [43]. Изучение 100 H.pylori-серопозитивных пациентов выявило у большинства из них аутоантитела, перекрестно реагировавшие с желудочными антигенами. Активность аутоантител определялась прежде всего в области эпителиоцитов желудочных желез, особенно вдоль апикального края главных и париетальных клеток, снижаясь в направлении от шеечных отделов к основаниям желез. При этом выявлялась корреляция между лимфоцитарной инфильтрацией в области шеечных эпителиоцитов и наличием аутоантител. Исследователи предположили, что антитела могут формироваться в результате перекрестной реакции с антигенными детерминантами муцина, высвобождающимися при регенерации желудочного эпителия. Важным является тот факт, что у пациентов с наличием аутоантител чаще обнаруживались атрофия желез и лимфоцитарная инфильтрация, чем у H.pylori-позитивных пациентов без аутоантител. То есть хроническая аутоиммунная агрессия против желудочного эпителия может способствовать развитию атрофии и метаплазии в СОЖ при хеликобактерном гастрите. У пациентов с более высокими титрами аутоантител выявлялись более выраженные морфологические изменения. Иными словами, степень выраженности антигенной мимикрии обусловливает прогрессирование атрофии. Однако в связи с тем, что распространенность атрофического гастрита в различных географических регионах неодинакова, маловероятно, что молекулярная мимикрия может полностью обусловить его развитие, и для выявления других факторов необходимы дальнейшие исследования [30].
Атрофия при рефлюксе желчи скорее всего является следствием повторного эрозирования СОЖ в результате ее повреждения желчными кислотами или лизолецитином в комбинации с кислотой. Возможно также, что рефлюкс желчи ускоряет развитие атрофии при H.pylori-ассоциированном гастрите. Атрофия может быть также следствием длительно существующего химического или реактивного гастрита. Следовательно, нестероидные противовоспалительные средства и другие этиологические факторы (избыток поваренной соли, острая пища, алкоголь и курение) могут действовать независимо или чаще синергично с H.pylori-инфекцией, вызывая развитие атрофии и КМ. Защитное действие против развития атрофии СОЖ могут оказывать такие алиментарные факторы, как витамины С, Е, β-каротин [13, 25].
Таким образом, существуют два главных механизма развития атрофии СОЖ. Первый механизм обусловлен повреждением пролиферативного компартмента и/или деструкцией желез по-средством как прямого бактериального воздействия, так и воспалительного ответа организма хозяина. Другой — опосредован аутоиммунными реакциями, которые вызывают постепенное разрушение железистых эпителиоцитов с сохранением стволовых клеток.
Природа кишечной метаплазии
Под метаплазией понимают превращение одной разновидности ткани в другую, отличную от первой морфологически и функционально при сохранении ее основной видовой принадлежности. Метаплазия представляет собой неопухолевое изменение клеточного фенотипа, которое обычно возникает в ответ на персистирование неблагоприятных условий окружающей среды. Измененный фенотип является следствием соматических мутаций стволовых клеток или эпигенетических изменений, приводящих к нарушенной дифференцировке клеток потомства. Последующее возникновение измененного фенотипа в качестве доминирующей популяции является результатом селекционного давления, оказываемого измененной микросредой [14]. Таким образом, метаплазию можно рассматривать как потенциально обратимое изменение, при котором дифференцированные типы клеток замещаются другими дифференцированными типами клеток, обычно лучше приспособленных к изменившимся условиям среды. В то же время метаплазия нарушает нормальную функцию ткани и делает возможным дальнейшее ее преобразование в опухоль.
В СОЖ КМ представляет собой переход от желудочного эпителиального фенотипа к тонко- или толстокишечному эпителиальному фенотипу. Традиционно КМ разделяется на полную и неполную. Полная КМ (тонкокишечная), или тип I, характеризуется появлением клеток Панета и бокаловидных клеток, продуцирующих сиаломуцины, которые характерны для тонкокишечной слизистой. Неполная КМ включает II и III типы, которые характеризуются наличием призматического эпителия и бокаловидных клеток, продуцирующих сиаломуцины и/или сульфомуцины. При II типе метаплазии секретируются нейтральные или кислые сиаломуцины, а при III типе — сульфомуцины. При III типе КМ (толстокишечной метаплазии) отмечаются расширение желез и отсутствие клеток Панета [7].
В последние годы появились обоснованные суждения о том, что отождествлять понятия относительно КМ «тонкокишечная — полная» и «толстокишечная — неполная» не совсем корректно. Это объясняется тем, что даже при полной метаплазии не обнаруживается весь спектр клеток, характерных для тонкой кишки, а также нередкой встречаемостью смешанного типа КМ. С позиций современного понимания процесса метаплазии полную метаплазию можно рассматривать в качестве начального этапа такой перестройки, а неполную — как нарушение процессов дифференцировки в данной клеточной линии (рис. 2). Таким образом, понятия «полная метаплазия» и «неполная метаплазия» характеризуют полноту воспроизведения кишечного фенотипа клеток, а термины «тонкокишечная» и «толстокишечная» — отражают специализацию.
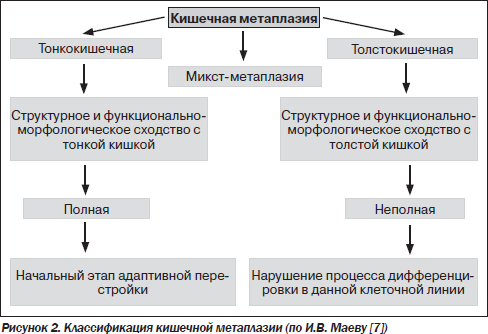
Полная КМ не имеет морфологических сходств с дисплазией, отчасти поэтому данный тип не относят к предраковым изменениям. Кроме того, тонкокишечный эпителий, образующий участки метаплазии, вообще мало склонен к малигнизации. Так, широко известным фактом является большая редкость рака тонкой кишки. О предраковом характере неполной КМ свидетельствует наличие в ней лактоферрина, который играет важную роль в обмене железа [41]. Как известно, железо необходимо для синтеза ДНК и соответственно в повышенном количестве требуется для репликации опухолевых клеток. Регуляция пролиферации и апоптоза в эпителиоцитах при толстокишечной метаплазии сильно нарушена, в некоторых из них выявляется мутация гена р53, что позволяет данным клеткам подвергаться дальнейшему перерождению под влиянием различных мутагенов, вплоть до злокачественного, поскольку они защищены от апоптоза [17]. Существуют данные о том, что при III типе КМ вероятность развития рака желудка в 4 раза выше, чем при I типе [42]. Наличие неполной КМ показало высокую специфичность этого признака (98 %) для РЖ, однако чувствительность оказалась достаточно низкой — всего 38 % [38]. То есть неполная КМ не может рассматриваться как маркер прогноза развития РЖ кишечного типа. Высказывается мнение, что маркером повышенного риска возникновения РЖ является не столько тип КМ, сколько площадь замещения желудочного эпителия [27]. Это обусловлено возможностью абсорбировать и депонировать очагами полной КМ ряда потенциальных канцерогенов в желудке. Таким образом, важнейшими факторами, способствующими развитию РЖ у больных КМ, являются неполный ее тип и большая площадь замещения желудочного эпителия [26].
Желудочная эпителиальная дисплазия
Термин «желудочная эпителиальная дисплазия» предполагает наличие эпителия с выраженными клеточными и структурными аномалиями и высокой склонностью к неопластической трансформации, независимо от наличия или отсутствия метаплазии. Природа дисплазии до сих пор не выяснена. Участки с эпителиальной дисплазией могут обнаруживаться в окружности участков РЖ кишечного типа в некоторых, но не во всех случаях. Это заставляет предположить возможность прогрессии дисплазии в карциному — подобно развитию колоректального рака из аденомы. Дисплазия эпителия, особенно тяжелая, рассматривается как облигатное явление на этапе существования рака in situ и обычно расценивается патологами как предраковое изменение [45].
До недавнего времени существовали значительные различия в интерпретации неопластических изменений желудка японскими и западноевропейскими морфологами. Дисплазия высокой степени, диагностированная европейскими, японскими исследователями, рассматривалась как неинвазивный рак [4,16]. С целью унификации оценки дисплазии в 1998 сначала была принята Падуанская классификация желудочной дисплазии, а затем в этом же году в Вене группа ведущих морфологов выработала Консенсус по классификации опухолей желудочно-кишечного тракта. Этот Консенсус стал известен как Венская классификация желудочно-кишечной эпителиальной неоплазии [35, 36]:
Категория 1. Отсутствие неоплазии/дисплазии.
Категория 2. Неопределенность относительно неоплазии/дисплазии.
Категория 3. Неинвазивная неоплазия низкой степени (аденома/дисплазия низкой степени).
Категория 4. Неинвазивная неоплазия высокой степени.
4.1. Высокая степень аденомы/дис-плазии.
4.2. Неинвазивный рак (carcinoma in situ).
4.3. Подозрение на инвазивный рак.
Категория 5. Инвазивная неоплазия.
5.1. Внутрислизистый рак.
5.2. Рак с распространением на подслизистый слой или глубже.
Венская классификация предопределяет стратегию дальнейшего клинического ведения пациента. Категория 1 (отсутствие неоплазии или дисплазии) включает нормальную слизистую оболочку, а также слизистую с реактивными, регенеративными, гиперпластическими, атрофическими и метапластическими изменениями. Дальнейшее обследование может быть необходимо или нет. Категория 2 (неопределенность в отношении неоплазии или дисплазии) требует проведения последующих исследований для выяснения истинной природы поражения. При категории 3 неоплазия присутствует, однако риск возникновения инвазивного рака низкий, поэтому рекомендуется местное лечение поражения в настоящее время или проведение наблюдения. В случае категории 4 существует риск инвазии и роста опухоли, поэтому больным показана эндоскопическая резекция слизистой оболочки или хирургическое лечение. Больным категории 5 показано срочное оперативное лечение в связи с опасностью дальнейшего роста опухоли и метастазирования. При использовании Венской классификации совпадение патоморфологических заключений японских и западноевропейских специалистов возросло примерно в 2 раза. Однако в пояснении к Венской классификации указывается, что данная градация дисплазий важна в первую очередь для исследовательских целей, а не для клинической практики.
В практической работе преимущественно используют двухстадийное разделение дисплазии на низкую и высокую степени. При этом обнаружение у пациента дисплазии высокой степени считается облигатным предраком и является показанием к хирургическому лечению (эндоскопической мукозэктомии или резекции желудка). Выявление дисплазии низкой степени требует тщательного пересмотра несколькими патологами и динамического наблюдения за пациентом [10].
Классификация ХГ
На всех этапах изучения ХГ существовали классификации, которые отражали научные знания о заболевании в данном историческом отрезке времени. Новейшая история классификаций ХГ берет начало с Сиднейской системы (1990 г.) и ее Хьюстонского усовершен-ствования (1996 г.) [24]. В соответствии с этой системой выделяются три типа гастрита (неатрофический, атрофический и особые формы), указываются локализация процесса (антрум, тело и тотальное поражение), этиологический фактор (наиболее частой причиной является H.руlоri) и морфологическая характеристика (табл. 1).
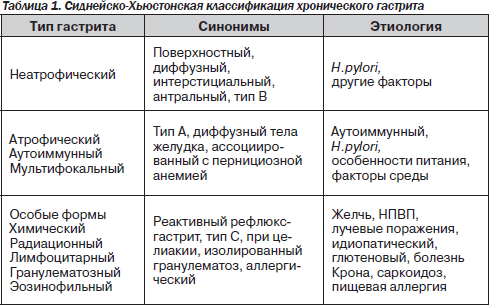
Для проведения морфологического исследования рекомендовано исследование 5 гастробиоптатов (1 из угла желудка, 2 из тела и 2 из антрума), в которых описывают основные патоморфологические изменения: хроническое воспаление (инфильтрация клетками лимфоплазмоцитарного ряда), активность (нейтрофильная инфильтрация), атрофию, кишечную метаплазию (полная, неполная), дисплазию. Наличие и выраженность каждого из указанных признаков описываются в баллах в соответствии с 4-уровневой визуально-аналоговой шкалой (0 — отсутствие признака, 1 балл — незначительное проявление признака, 2 балла — умеренное, 3 балла — выраженное). Необходимо отметить, что именно Сиднейская классификация и ее Хьюстонская модификация официально приняты в большинстве стран, в том числе и в Украине [10].
В рамках Сиднейской классификации предложено выделять два фенотипа хеликобактерного гастрита — классический антральный и фундальный (мультифокальный). Именно топографические особенности гастрита, а не выраженность воспаления, определяют клинические последствия инфицирования H.pylori. Установлено, например, что у 1 % больных антральным хроническим гастритом ежегодно возникают дуоденальные язвы, но у них не развивается рак желудка. При фундальном и мультифокальном гастритах у 1 % пациентов ежегодно развивается РЖ и практически не встречаются дуоденальные язвы. В связи с этим предложено выделять «язвенный» (патологические изменения локализуются только в антруме) и «раковый» (поражаются антрум и тело желудка) фенотипы ХГ [3].
Возможное объяснение указанным взаимосвязям кроется в том, что фенотип H.pylori-accoцииpoванного гастрита влияет на секрецию соляной кислоты. Если ее уровень низкий, H.pylori может колонизировать любой отдел желудка, при сохранной (повышенной) кислотности единственным местом, где может выжить микроорганизм, является антральный отдел, для которого характерны более высокие значения рН. В этом случае ведущую роль в развитии конкретного фенотипа будет играть возраст, в котором произошло заражение, поскольку для детей более характерно состояние гипоацидности, а для взрослых — нормацидности [17].
При классификации ХГ ключевым вопросом является оценка возможного риска развития РЖ у конкретного пациента. В 2002 г. Международная группа по изучению атрофии, в которую входили такие авторитетные ученые, как M. Rugge, P. Correa, M.F. Dixon, предложила выделять два основных типа атрофии: метапластический и неметапластический (рис. 3) [34].
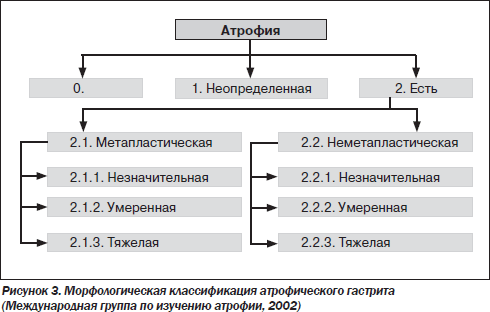
Неметапластический тип характеризуется потерей желез, сопровождающейся фиброзом или фибромускулярной пролиферацией собственной пластинки слизистой оболочки. При метапластическом типе наблюдается замещение обычных желез метапластическими (кишечными), что может иметь место на фоне других признаков атрофии. Как метапластическая, так и неметапластическая атрофия должна иметь характеристику по трем степеням тяжести в соответствии с Сиднейской системой. Обычно морфологическими критериями тяжелой атрофии считают потерю более 60 % желез, умеренной — потерю 30–60 % желез и легкой — менее 30 % желез [1, 34].
В 2008 г. группа экспертов, в которую вошли известные патологоанатомы, занимающиеся патологией желудка, и ряд гастроэнтерологов-клиницистов, предложили новую систему оценки гастрита — систему OLGA (Operative Link for Gastritis Assessment). В этой системе применяется оценка гистологической выраженности атрофии и воспаления в антральном отделе (3 биоптата) и в теле желудка (2 биоптата) с последующим определением интегральных показателей — стадии и степени хронического гастрита (табл. 2, 3) [32, 33].
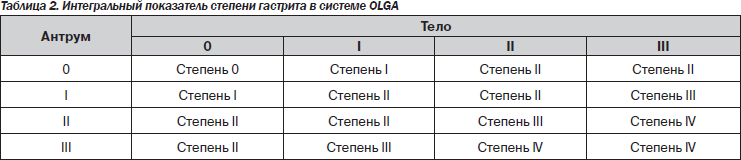
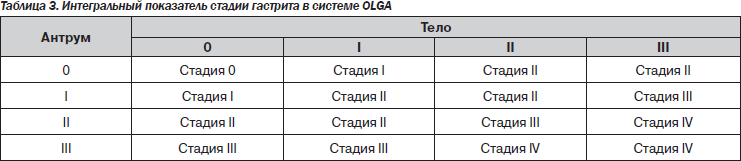
Под степенью гастрита подразумевается выраженность суммарной воспалительной инфильтрации (нейтрофильными лейкоцитами и мононуклеарными клетками), под стадией — выраженность атрофии. Такая система должна дать достаточно полную характеристику гастрита и отразить его динамику. Имеется в виду, что риск рака тем выше, чем более выражена атрофия и чем больше объем поражения. Пациенты с III и IV стадиями атрофии относятся к группе высокого риска развития некардиального РЖ. В этой же работе была предложена новая визуально-аналоговая шкала определения стадии гастрита.
Система OLGA, на наш взгляд, имеет два существенных преимущества по сравнению с предыдущими классификациями. Во-первых, определение стадии атрофии позволяет стратифицировать риск развития у пациента РЖ, а во-вторых, интегральный подход позволяет объективно определить наличие и выраженность регрессирования степени воспаления и стадии атрофии в результате лечения.
Серологическая диагностика при заболеваниях гастродуоденальной зоны
Одна из проблем диагностики атрофии СОЖ заключается в очаговости процесса. Не вызывает сомнений тот факт, что даже 5 биоптатов из разных отделов Ж не могут отразить состояния СО целиком. Объективность гистологического заключения несколько повышается при заборе биопсионного материала во время проведения хромоэндоскопии. Однако и в этом случае мы можем судить о состоянии СО лишь по изменениям в нескольких биоптатах. Для преодоления указанных диагностических особенностей финскими учеными была предложена серологическая диагностика патологических изменений в СОЖ.
Предложенный метод — «гастропанель» — основан на определении в сыворотке крови уровня гастрина-17, пепсиногена I и II и антител IgG к H.pylori. Гастрин-17 вырабатывается G-клетками антрального отдела желудка, и при атрофии в этом отделе уровень гастрина снижается и составляет менее 5 пмоль/л. В случае атрофического гастрита тела желудка при отсутствии атрофии в антруме содержание гастрина-17 возрастает (> 10 пмоль/л) в связи с включением механизма отрицательной обратной связи регуляции кислото-продукции через гастрин. При атрофии тела желудка снижается уровень сывороточного пепсиногена-I (< 25 мкг/л), который продуцируется главными клетками тела желудка. Пепсиноген-II вырабатывается во всех отделах желудка и в двенадцатиперстной кишке, и при атрофическом гастрите тела желудка снижается отношение пепсиноген-I/пепсиноген-II. Повышение уровня антител IgG к H.pylori до 38 ЕIU и более свидетельствует о наличии хеликобактерной инфекции и соответственно хеликобактерного гастрита. В отличие от морфологической серологическая диагностика ХГ интегративно отражает состояние СОЖ и фактически позволяет осуществлять неинвазивную скрининговую диагностику атрофического гастрита. Разумеется, в дальнейшем пациентам с признаками атрофии по серологическим данным следует провести эндоскопию с гастробиопсией для верификации патологии и получения детальной гистологической характеристики [18, 21].
Принципы лечения хронического гастрита
Терапия хронического гастрита осуществляется дифференцированно, в зависимости от клиники, этиопатогенетической и морфологической формы заболевания, кислотообразующей функции желудка. Безусловно, стратегическим направлением лечения ХГ является эрадикация H.pylori. В нашей стране получили признание Маастрихтские рекомендации по лечению заболеваний, ассоциированных с H.pylori. Важным аспектом в выборе терапии является учет резистентности H.pylori к метронидазолу и кларитромицину в конкретном регионе. К сожалению, в Украине нет статистических данных относительно этого вопроса, а в своей работе мы используем данные российских исследователей, предполагая, что в Украине схожая ситуация. Так, в России резистентность H.pylori к метронидазолу составляет более 40–45 %, а к кларитромицину — менее 6–8 %, к амоксициллину резистентность не обнаружена. В связи с указанным обстоятельством как в России, так и в Украине не рекомендовано использовать метронидазол в схемах антихеликобактерной терапии. Маастрихтским консенсусом III-2005 предложено продлить сроки лечения до 10–14 дней, однако допускается и 7-дневная терапия в тех регионах, где она дает хороший эффект [11, 15]. В качестве первой линии антихеликобактерной терапии рекомендовано использовать как тройную, так и квадротерапию с добавлением субцитрата висмута. Полученные нами данные указывают на то, что 7-дневная квадротерапия (ИПП, амоксициллин, кларитромицин и висмута трикалия дицитрат) обеспечивает уровень эрадикации более 90 %. Кроме этого, 7-дневная квадротерапия по сравнению с 10- и 14-дневными тройными схемами лучше переносится больным. На наш взгляд, в виде первой линии вообще не стоит использовать 14-дневную терапию, а следует оставить ее в качестве резерва [10].
Хронический антральный гастрит, Н.pylori-ассоциированный (тип В)
Для лечения используются две линии антихеликобактерной терапии: начинают лечение с первой линии, в случае неудачи назначают вторую линию.
Первая линия антихеликобактерной терапии
Первый вариант. ИПП в стандартной дозе 2 раза в день, амоксициллин 1000 мг 2 раза в день, кларитромицин 500 мг 2 раза в день — в течение 10 дней.
Второй вариант (четырехкомпонент- ная терапия). ИПП в стандартной дозе, амоксициллин 1000 мг 2 раза в день, кларитромицин 500 мг 2 раза в день — 7 дней и висмута трикалия дицитрата 240 мг 2 раза в день продолжительностью 14 дней. Дополнительное использование висмута трикалия дицитрата не только позволяет повысить степень эрадикации Н.pylori, но и способствует более быстрому разрешению нейтрофильной и лимфоплазмоцитарной инфильтрации СОЖ. Кроме этого, в данной схеме длительность антибактериальной терапии составляет 7 дней, а не 10 или 14 дней, поэтому она может быть рекомендована лицам, которые в прошлом отмечали плохую переносимость и/или аллергические реакции при применении антибиотиков.
Третий вариант (при наличии атрофии слизистой оболочки желудка с ахлоргидрией, подтвержденной при рН-метрии). Амоксициллин 1000 мг 2 раза в день, кларитромицин 500 мг 2 раза в день, висмута трикалия дицитрат 240 мг 2 раза в день продолжительностью 10–14 дней.
Индивидуальные особенности антихеликобактерной терапии. У пациентов, которые указывают на аллергические реакции, непереносимость полусинтетических пенициллинов или макролидов, у пожилых больных, у пациентов с нарушенной функцией печени и/или почек, в ситуациях, при которых полноценная антихеликобактерная терапия невозможна, используют следующий вариант лечения: ИПП в стандартной дозировке, один антибиотик, который переносит пациент, в стандартной дозе, висмута трикалия дицитрат 240 мг 2 раза в день продолжительностью 10–14 дней.
Вторая линия антихеликобактерной терапии
Проводится при отсутствии эрадикации Н.pylori после лечения больных одним из вариантов терапии первой линии.
Первый вариант (используется при условии, что пациент ранее не принимал орнидазол). ИПП в стандартной дозировке, амоксициллин 1000 мг 2 раза в день (к амоксициллину практически отсутствует резистентность), висмута трикалия дицитрат по 240 мг 2 раза в день, орнидазол по 500 мг 3 раза в день — 14 дней.
Второй вариант. ИПП в стандартной дозировке, амоксициллин 1000 мг 2 раза в день, нифурател (макмирор) (400 мг 2 раза в день) или фуразолидон (100 мг 4 раза в день), висмута трикалия дицитрат 240 мг 2 раза в день продолжительностью 14 дней.
NB! Контроль эрадикации рекомендуется проводить неинвазивными методами для исключения реинфицирования во время проведения гастродуоденоскопии.
Что делать, если эрадикация не наступила после второй линии лечения?
При отсутствии эрадикации Н.pylori после лечения препаратами второй линии рекомендуется подбор терапии только после определения чувствительности Нр к антибиотикам.
Особенности ведения пациентов с хроническим атрофическим гастритом
После эрадикации H.pylori (обязательное условие) пациентам 1 раз в 6 месяцев проводят курс лечения висмута трикалия дицитратом 240 мг 2 раза в день продолжительностью 28 дней. Применение висмута кроме антихеликобактерного и противовоспалительного эффекта способствует стабилизации процессов апоптоза и пролиферации в слизистой оболочке желудка, обеспечивает антиоксидантный эффект.
Кроме этого, пациентам с хроническим атрофическим гастритом рекомендованы антиоксиданты: церулоплазмин 10 мл в/в № 10, мелатонин 6 мг в сутки, α-токоферол 400–800 МЕ в сутки — 3–6 месяцев, и другие препараты из этой группы, прием которых необходимо чередовать между собой.
У пациентов со сниженной кислотообразующей функцией желудка и при ахлоргидрии с заместительной целью используют пепсидил по 1–2 столовых ложки 3 раза в день (во время еды) или плантаглюцид по 0,5–1,0 г (по 1/2–1 чайной ложке) 2–3 раза в день за 20–30 мин до еды. Назначают панкреоферменты (креон, мезим форте, пангрол, панзинорм) в индивидуально подобранной дозе. Длительность заместительной терапии зависит от тяжести состояния пациента.
Мониторинг пациентов с атрофическим гастритом. Пациентам с хроническим атрофическим гастритом стадии I и II (по системе OLGA) необходимо проводить эндоскопический и морфологический мониторинг один раз в год, а при атрофии III и IV стадий — один раз в 6 месяцев.
Лечение рефлюкс-гастрита (тип С)
Причиной рефлюкс-гастрита является заброс (рефлюкс) дуоденального содержимого в желудок. При дуоденогастральном рефлюксе повреждающее воздействие на СОЖ оказывают желчные кислоты и лизолецитин. Повреждающие свойства желчных кислот зависят от рН желудка: при рН < 4 наибольшее воздействие на СО оказывают тауриновые конъюгаты, а при рН > 4 — неконъюгированные желчные кислоты, обладающие значительно большим повреждающим действием.
При лечении рефлюкс-гастрита используют:
— висмута трикалия дицитрат (120 мг 4 раза или 240 мг 2 раза в день);
— сукральфат (500–1000 мг 4 раза в день) — наиболее эффективно связывает конъюгированные желчные кислоты при рН 2, при повышении рН этот эффект снижается, поэтому нецелесообразно его одновременное назначение с антисекреторными препаратами;
— препараты урсодезоксихолевой кислоты (10 мг/кг/сут) в течение 1–1,5 месяца;
— для нормализации моторной функции прокинетики (метоклопрамид, домперидон) и регуляторы моторики желчного пузыря (мебеверин).
Заключение
Современная диагностика ХГ направлена на определение этиологического фактора и характера поражения СОЖ. Новая классификация гастритов — система OLGA — позволяет стратифицировать риск развития РЖ. Использование «гастропанели» в комплексе с морфологическим исследованием позволяет расширить наши представления о структурно-функциональном состоянии желудка. Современные подходы к лечению строго дифференцированы, воздействуют на этиологию и патогенез каждой конкретной формы ХГ, не только позволяют предотвратить развитие язвенной болезни и РЖ, но и способствуют регрессу патологических процессов в СОЖ.
1. Аруин Л.И., Кононов А.В., Мозговой С.И. Новая Классификация хронического гастрита // Актуальные вопросы патологической анатомии: Материалы III съезда Рос. общества патологоанатомов. — Самара, 2009. — Т. 1. — С. 5-8.
2. Аруин Л.И. Helicobacter pylori: каким образом один возбудитель вызывает разные болезни // Эксперим. и клин. гастроэнтерол. — 2004. — № 1. — C. 36-41.
3. Аруин Л.И. Из 100 инфицированных Helicobacter pylori рак желудка возникает у двоих. Кто они? // Эксперим. и клин. гастроэнтерол. — 2004. — № 1 (внеочередной вып.). — С. 12-18.
4. Бабак О.Я. Современные представления об оценке риска развития и профилактике рака желудка // Сучас. гастроентерологія. — 2009. — № 6. — С. 62-66.
5. Барышникова Н.В., Успенский Ю.П., Ткаченко Е.И. Оптимизация лечения больных с заболеваниями, ассоциированными с инфекцией Helicobacter pylori: обоснование необходимости использования препаратов висмута // Эксперим. и клин. гастроэнтерол. — 2009. — № 6. — С. 116-121.
6. Ивашкин В.Т., Шептулин А.А., Лапина Т.Л. Хронический гастрит, вызванный инфекцией Helicobacter pylori: диагностика, клиническое значение, прогноз: Пособие для врачей. — М.: РГА, 2009. — 23 с.
7. Маев И.В., Зайратьянц О.В., Кучерявый Ю.А. Кишечная метаплазия слизистой оболочки желудка в практике гастроэнтеролога: современный взгляд на проблему // Рос. журн. гастроэнтерол., гепатол., колопроктол. — 2000. — № 4. — С. 38-48.
8. Пасечников В.Д., Котелевец С.М., Чуков С.З. Морфофункциональные проявления атрофии слизистой оболочки желудка при H.pylori-ассоциированном гастрите // Рос. журн. гастроэнтерол., гепатол., колопроктол. — 2004. — Т. 14, № 1. — С. 26-32.
9. Пасечников В.Д., Чуков С.З. Эпидемиология рака желудка // Рос. журн. гастроэнтерол., гепатол., колопроктол. — 2002. — Т. 12, № 3. — С. 18-26.
10. Островський О.С., Мосійчук Л.М., Зак М.Ю., Гайдар Ю.А. Особливості морфологічних та гістотопографічних змін в слизовій оболонці шлунка у пацієнтів з хронічним атрофічним гастритом при хелікобактерній інфекції // Гастроентерологія: Міжвід. зб. — Дніпропетровськ, 2009. — Вип. 42. — С. 232-241.
11. Стандарты диагностики и лечения кислотозависимых и ассоциированных с Helicobacter pylori заболеваний (четвертое Московское соглашение. Приняты X съездом НОГР 5 марта 2010 г.). — М., 2010. — 10 с.
12. Циммерман Я.С. Клиническая гастроэнтерология. — М.: Гэотар-Медиа, 2009. — 416 с.
13. Чернин В.В. Болезни пищевода, желудка и двенадцатиперстной кишки: Руководство для врачей. — М.: Мед. информ. агентство, 2010. — 528 с.
14. Чернин В.В. Хронический гастрит. — Тверь: Триада, 2006. — 304 с.
15. Шептулин А.А., Киприанис В.А. Диагностика и лечение инфекции Helicobacter pylori: основные положения согласительного совещания «Маастрихт-3»: по материалам Всемирного конгресса гастроэнтерологов в Монреале и Европейской недели гастроэнтерологов в Копенгагене // Рос. журн. гастроэнтерол., гепатол., колопроктол. — 2006. — № 2. — С. 88-91.
16. Филиппов Ю.А. Рак желудка. Ранняя диагностика и лечение // Гастроентерологія: Міжвід.зб. — Вип. 38. — 2007. — С. 307-315.
17. Bartnik W. Clinical aspects of Helicobacter pylori infection // Pol. Arch. Med. Wewn. — 2008, Jul — Aug. — Vol. 118(7–8). — Р. 426-430.
18. Copps J., Murphy R.F., Lovas S. The production and role of gastrin-17 and gastrin-17-gly in gastrointestinal cancers. // Protein Pept. Lett. — 2009. — Vol. 16(12). — Р. 1504-1518.
19. Correa P. A human model of gastric carcinogenesis // Cancer Res. — 1988. — Vol. 48. — P. 3554-3560.
20. Correa P. Helicobacter pylori and gastric cancer: state of the art // Cancer Epidemiol. Biomarkers Prew. — 1996. — Vol. 5. — P. 477-481.
21. Di Mario F., Cavallaro L.G. Non-invasive tests in gastric disease // Dig Liver Dis. — 2008. — Vol. 40(7). — Р. 523-530.
22. Dixon M.F., Genta R.M., Yardley J.H. Classification and grading of gastritis. The updated Sydney system. International Workshop on the Histopathology of gastritis, Houston 1994 // Am. J. Surg. Pathol. — 1996. — Vol. 20. — P. 1161-1181.
23. Dicksved J., Lindberg M., Rosenquist M. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls // J. Med. Microbiol. — 2009, Apr. — Vol. 58(Pt 4). — Р. 509-516.
24. El-Zimaity H. Gastritis and gastric atrophy // Curr Opin Gastroenterol. — 2008, Nov. — Vol. 24(6). — Р. 682-686.
25. Eussen S.J., Vollset S.E., Hustad S. Vitamins B2 and B6 and genetic polymorphisms related to one-carbon metabolism as risk factors for gastric adenocarcinoma in the European prospective investigation into cancer and nutrition // Cancer Epidemiol. Biomarkers Prev. — 2010, Jan. — Vol. 19(1). — P. 28-38.
26. Kodama M., Murakami K., Okimoto T. Guidelines for the management of Helicobacter pylori-Maastricht III-2005 and Japanese guidelines // Nippon Rinsho. — 2008, Apr. — Vol. 66(4). — Р. 804-810.
27. Konturek P.C., Konturek S.J., Brzozowski T. Helicobacter pylori infection in gastric cancerogenesis // J. Physiol. Pharmacol. — 2009, Sep. — Vol. 60(3). — Р. 3-21.
28. Liu Y.E., Gong Y.H, Sun L.P. Detection of Helicobacter pylori genotypes from human gastric mucosa and its association with gastric diseases // Zhonghua Yi XueZaZhi. — 2008, May. — Vol. 88(19). — Р. 1342-1346.
29. Marusawa H. Mechanisms of H. pylori infection-induced gastric carcinogenesis // GanTo Kagaku Ryoho. — 2010, Jan. — Vol. 37(1). — Р. 23-27.
30. Neesse A., Michl P., Barth P. Multifocal early gastric cancer in a patient with autoimmune atrophic gastritis and iron deficiency anaemia // Gastroenterol. — 2009, Feb. — Vol. 47(2). — Р. 223-227.
31. Proença-Modena J.L., Acrani G.O., Brocchi M. Helicobacter pylori: phenotypes, genotypes and virulence genes // Future Microbiol. — 2009, Mar. — Vol. 4(2). — Р. 223-240.
32. Rugge M., DE Boni M., Pennelli G. Gastritis OLGA-staging & gastric cancer risk: a twelve year clinico-pathological follow-up study // Minerva Gastroenterol. Dietol. — 2010, Mar. — Vol. 56(1). — P.13-17.
33. Rugge M., Correa P., Di Mario F. OLGA staging for gastritis: a tutorial // Dig Liver Dis. — 2008, Aug. — Vol. 40(8). — Р. 650-658.
34. Rugge M., Correa P., Dixon M.F. Gastric mucosal atrophy: interobserver consistency using new criteria for classification and grading // Aliment. Pharmacol. Ther. — 2002. — Vol. 16. — Р. 1249-1259.
35. Rugge M., Correa P., Dixon M.F. Gastric dysplasia. The Padova international classification // Am. J. Surg. Pathol. — 2000. — Vol. 24(2). — Р. 167-176.
36. Schlemper R.J., Riddell R.H., Kato Y. The Vienna classification of gastrointestinal epithelial neoplasia // Gut. — 2000. — Vol. 47. — Р. 251-255.
37. Sierra R., Une C., Ramirez V. Relation of atrophic gastritis with Helicobacter pylori-CagA(+) and interleukin-1 gene polymorphisms // World J Gastroenterol. — 2008, Nov. — Vol. 14(42). — Р. 6481-6487.
38. Stec-Michalska K., Peczek L., Michalski B. Helicobacter pylori infection and family history of gastric cancere decrease expression of FHIT tumor suppressor gene in gastric mucosa of dyspeptic patients // Helicobacter. — 2009, Oct. — Vol. 14(5). — Р. 126-134.
39. Suriani R., Colozza M., Cardesi E. CagA and VacA Helicobacter pylori antibodies in gastric cancer // Can. J. Gastroenterol. — 2008, Mar. — Vol. 22(3). — Р. 255-258.
40. Tanko M.N., Manasseh A.N., Echejoh G.O. Relation between Helicobacter pylori, inflammatory (neutrophil) activity, chronic gastritis, gastric atrophy and intestinal metaplasia // Niger J. Clin. Pract. — 2008, Sep. — Vol. 11(3). — Р. 270-274.
41. Torisu T., Matsumoto T., Takata Y. Atrophic gastritis, but not antibody to Helicobacter pylori, is associated with body mass index in a Japanese population // J. Gastroenterol. — 2008. — Vol. 43(10). — Р. 762-766.
42. Vannella L., Lahner E., Osborn J. Risk factors for progression to gastric neoplastic lesions in patients with atrophic gastritis // Aliment. Pharmacol. Ther. — 2010, Feb. — Vol. 13(2). — Р. 94-100.
43. Veijola L.I., Oksanen A.M., Sipponen P.I. Association of autoimmune type atrophic corpus gastritis with Helicobacter pylori infection // World J. Gastroenterol. — 2010, Jan. — Vol. 16(1). — Р. 83-88.
44. Vorobjova T., Maaroos H.I., Uibo R. Immune response to Helicobacter pylori and its association with the dynamics of chronic gastritis in the antrum and corpus // APMIS. — 2008, Jun. — Vol. 116(6). — Р. 465-476.
45. Yeh L.Y., Raj M., Hassan S. Chronic atrophic antral gastritis and risk of metaplasia and dysplasia in an area with low prevalence of Helicobacter pylori // Indian J. Gastroenterol. — 2009, Mar — Apr. — Vol. 28(2). — Р. 49-52.