
Международный эндокринологический журнал Том 19, №3, 2023
Вернуться к номеру
Синдром Клайнфельтера в поєднанні із сімейним чоловічим передчасним статевим дозріванням (клінічний випадок)
Авторы: Сорокман Т.В., Колєснік Д.І., Черней Н.Я.
Буковинський державний медичний університет, м. Чернівці, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
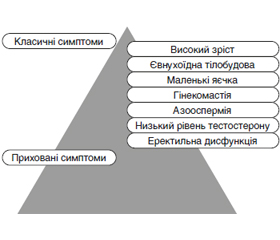
Автори наводять огляд літератури стосовно синдрому Kлайнфельтера в поєднанні із сімейним чоловічим передчасним статевим дозріванням і описують клінічний випадок. Синдром Клайнфельтера характеризується надзвичайною гетерогенністю клінічних і генетичних проявів. Поширеність синдрому Клайнфельтера становить від 0,1 до 0,2 % у новонароджених немовлят чоловічої статі та зростає до 3–4 % серед безплідних чоловіків і до 10–12 % у пацієнтів з азооспермією. На сьогодні невідомо, як лікувати пацієнтів з легким синдромом Клайнфельтера, який залишається недіагностованим або поєднується з іншою генетичною патологією, зокрема з гонадотропін-незалежним передчасним статевим дозріванням. Недостатня ефективність традиційної терапії хворих із синдромом Клайнфельтера та іншими видами генетично обумовленої секреторної безплідності ставить завдання пошуку нових методів лікування цієї патології. Сучасні досягнення медичної генетики, ендокринології, урології, репродуктології часто не дозволяють заповнити андрогенну недостатність, вилікувати ендокринну форму еректильної дисфункції та безплідність у цих пацієнтів. Це захворювання спричинене автосомно-домінантним успадкованим активуючим патогенним варіантом гена, що кодує рецептор лютеїнізуючого гормону/хоріогонадотропіну, який належить до сімейства G-білкових рецепторів. У чоловіків активація патогенних варіантів цього гена викликає надмірну секрецію тестостерону, що запускає раннє периферичне (передчасне) статеве дозрівання. Рекомендації щодо лікування розроблені частково головним чином через обмежену кількість зареєстрованих випадків, малий розмір вибірки й короткострокові результати. Поданий клінічний випадок є важливим з огляду на можливий ризик розвитку злоякісних новоутворень яєчок у пацієнтів із передчасним статевим дозріванням. Тому рекомендується тривале спостереження під час і після статевого дозрівання.
The article presents the results of a literature review on Klinefelter syndrome combined with familial male-limited precocious puberty and describes a clinical case. Klinefelter syndrome is a form of male hypogonadism, characterized by the presence of an extra X chromosome, small testes, seminiferous tubule dysgenesis, high levels of gonadotropin, low serum testosterone level, underdeveloped secondary sex characteristics and male infertility. Klinefelter syndrome is characterized by extreme heterogeneity of clinical and genetic manifestations. The prevalence of Klinefelter syndrome is 0.1 to 0.2 % in male newborns and increases to 3 to 4 % among infertile men and 10 to 12 % in patients with azoospermia. Currently, it is not known how to treat patients with mild Klinefelter syndrome that remains undiagnosed or is combined with other genetic pathology, including gonadotropin-independent precocious puberty. This disease is caused by an autosomal dominant inherited activating pathogenic variant of the gene encoding the luteinizing hormone/chorionic gonadotropin receptor, which belongs to the family of G protein-coupled receptors. In men, activation of pathogenic variants of this gene causes excessive secretion of testosterone, which triggers early peripheral (precocious) puberty. Treatment recommendations have been developed in part mainly because of the limited number of reported cases, small sample sizes, and short-term outcomes. The presented clinical case is important in view of the possible risk of developing malignant testicular neoplasms in patients with precocious puberty. Therefore, long-term follow-up during and after puberty is recommended. It is of great importance to take into account the aforementioned clinical manifestations in order to made early diagnosis of this syndrome, offer timely genetic counseling to parents, and rehabilitate these patients physically, psychically and socially.
синдром Клайнфельтера; сімейне чоловіче передчасне статеве дозрівання; клінічний випадок
Klinefelter syndrome; familial male-limited precocious puberty; clinical case
Для ознакомления с полным содержанием статьи необходимо оформить подписку на журнал.
- Butler G., Srirangalingam U., Faithfull J., Sangster P., Senniappan S., Mitchell R. Klinefelter syndrome: going beyond the diagnosis. Arch. Dis. Child. 2023. 108(3). 166-171. doi: 10.1136/archdischild-2020-320831.
- Verhoeven W.M.A., Egger J.I.M., Mergler S., Meijer T.A.A., Pfundt R., Willemsen M.H. A Patient with Moderate Intellectual Disa–bility and 49, XXXYY Karyotype. Int. J. Gen. Med. 2022. 15. 2799-2806. doi: 10.2147/IJGM.S348844.
- Thompson T., Howell S., Davis S. et al. Current survey of early childhood intervention services in infants and young children with sex chromosome aneuploidies. Am. J. Med. Genet. C. Semin. Med. Genet. 2020. 184(2). 414-427. doi: 10.1002/ajmg.c.31785.
- Simonetti L., Ferreira L.G.A., Vidi A.C. et al. Intelligence Quotient Variability in Klinefelter Syndrome Is Associated With GTPBP6 Expression Under Regulation of X-Chromosome Inactivation Pattern. Front. Genet. 2021. 12. 724625. doi: 10.3389/fgene.2021.724625.
- Kinjo K., Yoshida T., Kobori Y. et al. Random X chromosome inactivation in patients with Klinefelter syndrome. Mol. Cell. Pediatr. 2020. 7(1). 1. doi: 10.1186/s40348-020-0093-x.
- Gravholt C.H., Chang S., Wallentin M., Fedder J., Moore P., Skakkebæk A. Klinefelter Syndrome: Integrating Genetics, Neuropsychology, and Endocrinology. Endocr. Rev. 2018. 39(4). 389-423. doi: 10.1210/er.2017-00212.
- Tartaglia N., Ayari N., Howell S., D’Epagnier C., Zeitler P. 48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011. 100(6). 851-860. doi: 10.1111/j.1651-2227.2011.02235.x.
- Rajabzadeh M., Taheri N., Jazayeri O. 49,XXXXY syndrome: A case study and a systematic review of clinical features among the Iranian population. Clin. Case Rep. 2022. 10(9). e6342. doi: 10.1002/ccr3.6342.
- Hammami M.B., Elkhapery A. Sexual and developmental aspects of 49, XXXXY Syndrome: A case report. Andrologia. 2020. 52(10). e13771. doi: 10.1111/and.13771.
- Nassau D.E., Best J.C., Cohen J., Gonzalez D.C., Alam A., Ramasamy R. Androgenization in Klinefelter syndrome: Clinical spectrum from infancy through young adulthood. J. Pediatr. Urol. 2021. 17(3). 346-352. doi: 10.1016/j.jpurol.2021.02.021.
- Naotunna N.P.G.C.R., Liyanage C., Atapattu N. An infant with 49XXXXY syndrome: a case report. J. Med. Case Rep. 2021. 15(1). 630. doi: 10.1186/s13256-021-03188-4.
- Cornacchia M.A., Bhushan S., Arguello R. A Case of Familial Male-Limited Precocious Puberty in a Child With Klinefelter Syndrome. J. Endocr. Soc. 2018. 2(10). 1131-1136. doi: 10.1210/js.2018-00192.
- Topor L.S., Bowerman K., Machan J.T., Gilbert C.L., Kangarloo T., Shaw N.D. Central precocious puberty in Boston boys: A 10-year single center experience. PLoS One. 2018. 13(6). e0199019. doi: 10.1371/journal.pone.0199019.
- Cemeroglu A.P., Kaval D., Ozcan O. Etiology of Increased Referrals for Evaluation of Early Puberty in a Tertiary Care Center in Turkey: True Precocious Puberty, Obesity, or Parental Anxiety and Lack of Knowledge? Glob. Pediatr Health. 2021. 8. 2333794X211009096. doi: 10.1177/2333794X211009096.
- Goffredo M., Pilotta A., Parissenti I. et al. Early onset of puberty during COVID-19 pandemic lockdown: experience from two Pediatric Endocrinology Italian Centers. J. Pediatr. Endocrinol. Metab. 2023. 36(3). 290-298. doi: 10.1515/jpem-2022-0492.
- Ziqin L., Xiaohui L., Xiaobo C. Precocious Puberty in Boys: A Study Based on Five Years of Data from a Single Center in Northern China. J. Clin. Res. Pediatr. Endocrinol. 2021. 25. 13(4). 418-425. doi: 10.4274/jcrpe.galenos.2021.2021.0033.
- Gurnurkar S., DiLillo E., Carakushansky M. A Case of Familial Male-limited Precocious Puberty with a Novel Mutation. J. Clin. Res. Pediatr. Endocrinol. 2021. 13(2). 239-244. doi: 10.4274/jcrpe.galenos.2020.2020.0067.
- Qiao J., Han B. Diseases caused by mutations in lutenizing hormone/chorionic gonadotropin receptor. In: Tao YX (ed). Progress in molecular biology and translational science: G protein signaling pathways in health and disease. Cambridge, Academic Press, 2019. 69-89.
- Lane L.C., Flowers J., Johnstone H., Cheetham T. Adult height in patients with familial male-limited precocious puberty and the role of an aromatase inhibitor in patient management. J. Pediatr. Endocrinol. Metab. 2018. 31. 551-560. doi: 10.1515/jpem-2017-0363.
- Schedewie H.K., Reiter E.O., Beitins I.Z. et al. Testicular leydig cell hyperplasia as a cause of familial sexual precocity. J. Clin. Endocrinol. Metab. 1981. 52(2). 271-278. doi: 10.1210/jcem-52-2-271.
- Atta I., Laghari T.M., Khan Y.N., Lone S.W., Ibrahim M., Raza J. Precocious puberty in children. J. Coll. Physicians Surg. Pak. 2015. 25(2). 124-128.
- Aftab S., Manzoor J., Mahmood Q., Shaheen T. Precocious puberty: The clinical profile and the etiological classification of children presented at a tertiary care children’s hospital. Pak. J. Med. Sci. 2022. 38 (4 Part II). 955-959. doi: 10.12669/pjms.38.4.4816.
- Daussac A., Barat P., Servant N. et al. Testotoxicosis without Testicular Mass: Revealed by Peripheral Precocious Puberty and Confirmed by Somatic LHCGR Gene Mutation. Endocr. Res. 2020. 45(1). 32-40. doi: 10.1080/07435800.2019.1645163.
- Özcabi B., Tahmiscioğlu Bucak F., Ceylaner S. et al. Testotoxicosis: Report of Two Cases, One with a Novel Mutation in LHCGR Gene. J. Clin. Res. Pediatr. Endocrinol. 2015. 7(3). 242-248. doi: 10.4274/jcrpe.2067.
- Byambaragchaa M., Choi S.H., Kim D.W., Min K.S. Constitutive Activating Eel Luteinizing Hormone Receptors Induce Constitutively Signal Transduction and Inactivating Mutants Impair Bio–logical Activity. Dev. Reprod. 2021. 25(3). 133-143. doi: 10.12717/DR.2021.25.3.133.
- Arya V.B., Davies J.H. Idiopathic gonadotropin-independent precocious puberty — is regular surveillance required? J. Pediatr. Endocrinol. Metab. 2019. 32(4). 403-407. doi: 10.1515/jpem-2018-0419.
- Kremer H., Mariman E., Otten B.J. et al. Cosegregation of missense mutations of the luteinizing hormone receptor gene with familial male-limited precocious puberty. Hum. Mol. Genet. 1993. 2(11). 1779-1783. doi: 10.1093/hmg/2.11.1779.
- Cheuiche A.V., da Silveira L.G., de Paula L.C.P., Lucena I.R.S., Silveiro S.P. Diagnosis and management of precocious se–xual maturation: an updated review. Eur. J. Pediatr. 2021. 180(10). 3073-3087.doi: 10.1007/s00431-021-04022-1.
- Cattoni A., Albanese A. Case report: Fluctuating tumor mar–kers in a boy with gonadotropin-releasing hormone-independent precocious puberty induced by a pineal germ cell tumor. Front. Pediatr. 2022. 10. 940656. doi: 10.3389/fped.2022.940656.
- Kor Y. Central precocious puberty in a case of late-diagnosed familial testotoxicosis and long-term treatment monitoring. Hormones (Athens). 2018. 17(2). 275-278. doi: 10.1007/s42000-018-0029-1.
- Yuan X., Chen R., Zhang Y., Yang X., Lin X. Long-Term Treatment With Letrozole in a Boy With Familial Male-Limited Precocious Puberty. Front. Endocrinol. (Lausanne). 2022. 13. 906852. doi: 10.3389/fendo.2022.906852.
- Flippo C., Kolli V., Andrew M. et al. Precocious Puberty in a Boy With Bilateral Leydig Cell Tumors due to a Somatic Gain-of-Function LHCGR Variant. J. Endocr. Soc. 2022. 6(10). bvac127. doi: 10.1210/jendso/bvac127.
- Kooij C.D., Mavinkurve-Groothuis A.M.C., Kremer Hovinga I.C.L. et al. Familial Male-limited Precocious Puberty (FMPP) and Testicular Germ Cell Tumors. J. Clin. Endocrinol. Metab. 2022. 107(11). 3035-3044. doi: 10.1210/clinem/dgac516.
- Chang S., Skakkebaek A., Davis S.M., Gravholt C.H. Morbidity in Klinefelter syndrome and the effect of testosterone treatment. Am. J. Med. Genet. C. Semin. Med. Genet. 2020. 184(2). 344-355. doi: 10.1002/ajmg.c.31798.
- Sorokman T., Popeliuk N., Makarova O. Klinefelter syndrome in children and adolescents: combination of genetics and endocrinology. International Journal of Endocrinology (Ukraine). 2019. 15(3). 223-229. https://doi.org/10.22141/2224-0721.15.3.2019.172108.
- Tenedero C.B., Oei K., Palmert M.R. An Approach to the Evaluation and Management of the Obese Child With Early Puberty. J. Endocr. Soc. 2021. 6(1). bvab173. doi: 10.1210/jendso/bvab173.
- Fadich A., Giorgianni S.J., Rovito M.J. et al. USPSTF Testi–cular Examination Nomination-Self-Examinations and Examinations in a Clinical Setting. Am. J. Mens Health. 2018. 12(5). 1510-1516. doi: 10.1177/1557988318768597.
- Radmayr C., Dogan H.S., Hoebeke P. et al. Management of undescended testes: European Association of Urology/European Society for Paediatric Urology Guidelines [published correction appears in J. Pediatr. Urol. 2017. 13(2). 239. J. Pediatr. Urol. 2016. 12(6). 335-343. doi: 10.1016/j.jpurol.2016.07.014.