Вступ
На сьогодні лікування онкологічних захворювань є важливою соціально-економічною проблемою, яка кидає виклик системі охорони здоров’я всіх без виключення країн світу. За даними GLOBOCAN, у 2020 році в усьому світі виявлено близько 19,3 мільйона нових випадків та майже 10 мільйонів (9,9 млн) смертей від злоякісних пухлин [1]. Тому пошук нових та удосконалення вже існуючих методів лікування є актуальною проблемою сучасної онкології. Такими методами є різні класи імунотерапевтичних протипухлинних засобів, які впливають на формування імунної відповіді організму до клітин злоякісної пухлини. Основними класами імунотерапевтичних протипухлинних засобів є цитокіни (інтерлейкін-2, інтерферон), моноклональні антитіла, адаптивна Т-клітинна терапія та терапевтичні протиракові вакцини. Імунотерапія пухлин має багатовікову історію. Безліч повідомлень про спонтанний регрес злоякісних пухлин після перенесеного інфекційного захворювання та/або лихоманки з’явилися ще в Давньому Єгипті. Наступний етап розвитку протипухлинної терапії пов’язують з роботами Вільгельма Буша та Фрідріха Фелейзена, які в кінці ХІХ століття, незалежно один від одного, описали регресію пухлини у хворих після перенесеної бешихи. У 1868 році В. Буш навмисно заразив бешихою онкологічного хворого та зафіксував регресію пухлини. У 1882 році Ф. Фелейзен повторив експеримент В. Буша і в подальшому виділив збудника бешихи. Проте засновником імунотерапії злоякісних пухлин заслужено вважається Вільям Колі, який у 1893 році оприлюднив результати успішного лікування злоякісних пухлин повторним введенням вакцини на основі інактивованих збудників бешихи. Він створив так званий токсин Колі з вбитих Serratia marcescens та Streptococcus pyogenes, які значно провокували імунну відповідь проти пухлин. Наступний великий крок в імунотерапії стався в 1957 році, коли була проведена алогенна трансплантація гемопоетичних стовбурових клітин, що і досі успішно використовується для лікування гемобластозів. Подальшим кроком у розвитку стало лікування меланоми активованими імунними клітинами і цитокінами у кінці 1980-х років. Тоді ж терапію цитокінами дозволили для інших злоякісних новоутворень. Після відкриття В- і Т-лімфоцитів стався новий прорив у розвитку імунотерапії, який призвів до винайдення моноклональних антитіл. На сьогодні імунотерапія є ефективним методом лікування більшості злоякісних новоутворень, серед яких меланома, недрібноклітинний рак легень, рак печінки, шлунка, сечового міхура, шийки матки, деякі типи раку молочної залози, лімфоми тощо. Однак імунотерапія деяких злоякісних пухлин малоефективна, тому активно відбувається розробка нових та удосконалення існуючих засобів імунотерапії, і є сподівання, що показання до її застосування будуть розширюватися. З цією метою у даній роботі розглянуті принципи роботи різних класів імунотерапевтичних протипухлинних засобів, їх показання та можливі наслідки від застосування [2].
Використання цитокінів як протипухлинної імунотерапії
Цитокіни, такі як інтерферони, інтерлейкіни, лімфокіни, монокіни, хемокіни та фактори росту, є імунними модуляторами, що природно продукуються багатьма типами клітин. Вони являють собою білкові сигнальні молекули, що мають різноманітні фізіологічні функції, найбільш важливою серед яких є регуляція запалення та імунної відповіді [2]. Цитокіни здатні напряму стимулювати ефекторні та стромальні клітини в пухлинних вогнищах та посилювати розпізнавання пухлинних клітин цитотоксичними ефекторними клітинами. Саме тому системне застосування цитокінів (головним чином інтерферонів та інтерлейкінів) було одним із перших методів імунотерапії злоякісних пухлин. Ефективність цитокінів доведена в численних доклінічних дослідженнях з використанням багатьох моделей пухлин на тваринах. Зараз існує велика кількість досліджень із вивчення ефективності цитокінів, а саме гранулоцитарно-макрофагального колонієстимулюючого фактора (GM-CSF), інтерлейкіну-7, -12, -15, -18, -21 (IL-7, IL-12, IL-15, IL-18 і IL-21) у хворих із занедбаними формами злоякісних пухлин [3].
Лікування, засноване на цитокінах, стало можливим завдяки розробці технології рекомбінантної ДНК з використанням генно-інженерних штамів Escherichia coli. Це дозволило налагодити широкомасштабне виробництво очищених рекомбінантних людських цитокінів, придатних для системного введення пацієнтам. Управлінням з контролю якості харчових продуктів та лікарських засобів США (FDA) схвалено використання лікарських засобів на основі рекомбінантного інтерферону α (ІFN-α) та інтерлейкіну-2 (ІL-2) для лікування злоякісних пухлин, а також препарату іміквімод, який стимулює продукцію IL-12, IFN-γ та фактора некрозу пухлини (TNF) і використовується для локального лікування базальноклітинного раку шкіри [4].
Інтерферони — це цитокіни, які продукуються майже всіма клітинами організму та беруть участь у забезпеченні клітинного імунітету проти вірусних інфекцій. Клінічне застосування рекомбінантного ІFN-α для лікування злоякісних пухлин було схвалено FDA у 1986 році. ІFN-α є членом родини ІFN І типу, яке також включає ІFN-β, ІFN-σ, ІFN-κ та ІFN-ω. Механізм протипухлинної дії ІFN пов’язаний з його здатністю індукувати експресію молекул головного комплексу гістосумісності (МНС) І класу клітинами пухлин, стимулюванням дозрівання та/або активацією дендритних клітин (DC), природних кілерів (NK), цитотоксичних Т-лімфоцитів (CTL) та макрофагів. Крім цього, ІFN І типу здатні опосередковано стимулювати апоптоз пухлинних клітин та пригнічувати неоангіогенез пухлини [1, 4].
Природно інтерферони виділяються під час активації DAMP. Термін «молекулярні структури, пов’язані з небезпекою» (DAMP), описує як патогенні, так і непатогенні подразники, які активують патерн-розпізнавальні рецептори (PRR), щоб керувати вродженими імунними шляхами, що індукують інтерферони I та III типу та прозапальні цитокіни. Сімейство PRR включає Toll-подібні рецептори (TLR), які сприймають поверхневі патогенні та секреторні молекули, такі як бактеріальні ліпополісахариди (TLR1, TLR2, TLR4, TLR5 та TLR6) та нуклеїнові кислоти (TLR3, TLR7, TLR8 та TLR9). Також існують інші PRR, що сприймають вірусні нуклеїнові кислоти та інші ДНК. Неінфекційні подразники включають модифіковані компоненти позаклітинного матриксу, нуклеїнові кислоти або метаболіти, що виділяються з мертвих або відмираючих клітин ссавців, а також синтетичні сполуки, що імітують ці патогенні подразники. PRR виявлені в більшості клітин організму, де їх активація призводить до запуску механізмів, у результаті яких специфічні ферменти, такі як TANK-зв’язуюча кіназа 1 (TBK1), та IKKε та IκB активують фактор відповіді на ІFN та ядерний фактор κB (NF-κB) родини факторів транскрипції відповідно. Ці активовані фактори транскрипції сприяють експресії прозапальних цитокінів, таких як IL-6, TNF та ІFN-α, ІFN-β та ІFN-λ [5].
Інтерферони I типу активують протипухлинний імунітет за рахунок стимуляції вроджених та адаптивних популяцій цитотоксичних лімфоцитів (Т-клітини, NK, DC, вроджені лімфоїдні клітини) та пригнічення активності популяцій клітин, які пригнічують протипухлинний імунітет (наприклад, супресорні клітини мієлоїдного походження (MDSC) та Т-супресори). ІFN І типу також справляють внутрішній вплив на пухлинні клітини шляхом інгібування проліферації та модуляції апоптозу, диференціації, міграції та експресії антигену на клітинній поверхні. ІFN-α здатний викликати прямий каспазазалежний апоптоз пухлинних клітин. ІFN I типу можуть вироблятися як пухлинними, так і імунними клітинами, що призводить до активації імунних клітин, дія яких залежатиме від внутрішніх ефектів пухлинних клітин або доповнюватиме їх, включаючи презентацію антигену, продукцію цитокінів та шляхи передачі сигналів про апоптоз. Таким чином, вплив ІFN I типу на взаємні перехресні перешкоди між імунними та пухлинними клітинами є ключовим для їх протипухлинного потенціалу. Хоча конкретні механізми протипухлинного впливу ІFN точно не вивчені [2, 5], існує багато прикладів та відомих механізмів стійкості пухлинних клітин до впливу ІFN. Вони нагадують механізми, що використовуються вірусами, і впливають на різні рівні передачі сигналів IFN, включаючи видалення генів IFN типу I, зниження регуляції рецепторів та втрату сигнальних молекул, таких як STAT1 та IRF1 [5].
На основі більш глибокого аналізу продукції та дії IFN, а також впливу імуномодуляції на канцерогенез оновлено погляди щодо використання IFN для лікування злоякісних пухлин у певних умовах. Оскільки і імунні клітини, і самі клітини пухлини є потенційним джерелом IFN, взаємодії між пухлинною клітиною, IFN та імунними клітинами є ключем до підвищення протипухлинної ефективності терапії з використанням IFN (рис. 1).
/43.jpg)
Внутрішньопухлинна дія IFN на проліферацію, диференціацію, міграцію та антигенну презентацію доповнює дію активованих IFN імунних клітин на пухлину. Більша оцінка молекулярних сигнальних шляхів та наборів генів, активованих IFN, покращує здатність використовувати генні сигнатури відповідей на IFN для стратифікації пацієнтів. Для того, щоб розробити прогностичні маркери відповіді, провести цілеспрямовані клінічні випробування та розробити ефективні методи комбінованої терапії, необхідно зрозуміти справжні цілі та механізми дії терапії на основі IFN на конкретні підтипи раку. Майбутні дослідження повинні включати великі та цілеспрямовані імунологічні аналізи та збереження зразків, що дозволить розробити прогностичні стратегії IFN (які можуть показати деякі відмінності в ефективності між типами IFN), що полегшить підбір індивідуальних комбінацій. Якщо IFN проявляють протипухлинну дію через активацію протипухлинної імунної відповіді, можливо, вони не можуть бути ефективними при поширених формах захворювання у зв’язку з наявністю великої пухлинної маси та обумовленою мікросередовищем пухлини імунодепресією. Таким чином, терапія IFN може бути більш ефективною на ранніх стадіях лікування для знищення пухлинних клітин, які поширились за межі первинної пухлини і перебувають у кровоносних і/або лімфатичних судинах, що нагадує гемобластози, у випадку яких IFN показали себе більш ефективними. У цьому контексті ефективність все ще визначатиметься шляхом доставки відповідного IFN або індуктора IFN до клітини-мішені та мінімізації системної токсичності у наперед визначених пацієнтів, чутливих до даної терапії [5]. При терапії IFN майже у всіх пацієнтів спостерігаються деякі побічні ефекти, причому найбільш поширеними є втома, міалгія, нудота або блювання та діарея. Також частими є нервово-психічні побічні ефекти, такі як депресія та шкірні прояви, серед яких еритема, свербіж, рідше ліподистрофія, набряк та флебіт. Лабораторні побічні ефекти IFN включають гематологічні порушення, такі як лейкопенія, анемія та тромбоцитопенія, а також підвищення показників печінкових проб. Може виникати пізня автоімунна токсичність, що проявляється гіпотиреозом, васкулітом або гепатитом [6, 7].
Інтерлейкіни переважно секретуються CD4+ Т-лімфоцитами, які беруть участь у розвитку, активації та пригніченні CD8+ цитотоксичних Т-лімфоцитів, макрофагів та NK. З усіх інтерлейкінів найбільш детально вивчений IL-2. В 1970-х роках IL-2 був вперше охарактеризований як фактор росту Т-лімфоцитів та в подальшому був розроблений рекомбінантний –IL-2. Подібно до природного IL-2, рекомбінантний IL-2 активує імунну систему, сприяючи проліферації та диференціації NK, Т- та В-лімфоцитів. Крім того, рекомбінантний IL-2 підсилює цитолітичну активність лімфоцитів і сприяє взаємодії між злоякісними клітинами та імунною системою. У 1992 р. рекомбінантний IL-2 (альдеслейкін) отримав схвалення на лікування нирковоклітинного раку, а в 1998 р. було додано показання до метастатичної меланоми. Високі дози альдеслейкіну можуть використовуватися як окремий засіб, як частина багатокомпонентного режиму хіміоімунотерапії або як частина різноманітних підходів на основі клітинної імунотерапії раку. Рекомбінантний IL-2 є найбільш широко використовуваним інтерлейкіном для імунотерапії злоякісних пухлин. IL-2 є критичним кофактором, який активує цитотоксичні пухлиноінфільтруючі лімфоцити (TIL); посилює протипухлинну активність NK; індукує активовані лімфокіном клітини-кілери, які опосередковують протипухлинні ефекти; і сприяє зростанню та поширенню T-регуляторних клітин. Ключовою характеристикою IL-2 є залежність від дози, особливо ця залежність виражена в процесах активації різних підгруп імунних клітин. У високих дозах IL-2 стимулює імунну відповідь T-хелперів першого типу та сприяє протипухлинній активності цитотоксичних Т-лімфоцитів. Високі дози IL-2 викликають об’єктивну клінічну відповідь у 15–20 % та стійку повну регресію у 5–7 % хворих із занедбаними формами меланоми. Частота клінічної відповіді при внутрішньовенному болюсному введенні високих доз IL-2 у хворих на метастатичний нирковоклітинний рак становить близько 25 %, стійка повна регресія, аналогічно хворим на меланому, зафіксована у 7 % випадків. Тривалість клінічної відповіді становила від 24 до 54 місяців. У малих дозах IL-2 сприяє як посиленню, так і пригніченню імунної відповіді (стимулюючи Т-регуляторні клітини) [3]. Однак імунотерапія високими дозами IL-2 може спричинити гіпотензію, респіраторний дистрес, інтерстиціальний набряк легень та тромбоцитопенію. Також високі дози IL-2 можуть запускати фібриноліз і ДВЗ-синдром або, навпаки, бути причиною тромбозу (ТЕЛА, тромбоз вен печінки, мозку, серця), не пов’язаного з ДВЗ-синдромом [8, 9].
У клінічних випробуваннях вивчаються кілька інших рекомбінантних інтерлейкінів, включаючи рекомбінантні IL-7, IL-12, IL-15, IL-18, IL-21, IL-24 та інші. Різні складні білки, що містять інтерлейкіни, імуноцитокіни або фрагменти постійних складних білків, орієнтовані на конкретні рецептори інтерлейкінів або хемокінів, також використовуються в імунотерапії раку [2].
Загальні принципи взаємодії імунних клітин
Центральну роль у діяльності імунної системи відіграють лімфоцити. Існує два основних підтипи цих клітин — В- і Т-лімфоцити. В-лімфоцити розпізнають циркулюючі антигени в їх первинній формі й у відповідь на них виділяють захисні антитіла. Т-лімфоцити розпізнають пептидні антигени, які представлені на поверхні клітин унаслідок внутрішньоклітинної деградації.
Усі ядерні клітини організму містять на своїй поверхні МНС І типу. Частинки внутрішньоклітинних білків при розпаді виводяться і представляються на МНС І типу. Т-лімфоцити за допомогою Т-клітинних рецепторів (TCR) розпізнають антигени, приєднані до білків МНС. TCR — це клон-специфічний поверхневий білковий комплекс, що розпізнає антигени на МНС. Якщо клітина містить вірус, що порушує її роботу, або вона малігнізувалась, антигенний склад, представлений на МНС І типу, не відповідає нормальному, на що реагують CD8+ Т-лімфоцити (Т-цитотоксичні лімфоцити, CTL) і запускають пряму цитотоксичну реакцію, що призводить до загибелі цієї клітини. У свою чергу, антигенпрезентуючі клітини (макрофаги, дендритні клітини, моноцити, В-лімфоцити) експресують на своїй поверхні білки МНС ІІ типу, які виконують функцію представлення Т-лімфоцитам позаклітинних патогенів. Антигенпрезентуючі клітини фагоцитують патоген, внутрішньоклітинно проходить його лізис на менші пептиди, які представляються на МНС ІІ типу та взаємодіють з CD4+ Т-лімфоцитами (Т-хелпери). Існує підклас Т-клітин CD4+ CD25+ — Т-регуляторні клітини, що здатні пригнічувати імунну відповідь [10].
TCR на Т-клітинах проходить специфічне «навчання» в тимусі — позитивну і негативну селекцію. Ці процеси дозволяють допустити в загальний кровотік тільки ті клітини, що ефективно реагують на патогени і малігнізовані клітини, але є толерантними до власних антигенів [11]. Перш ніж Т-клітина почне виконувати свої функції, потрапивши в системний кровотік після селекції в тимусі, вона має пройти активацію. Для цього наївний Т-лімфоцит має зв’язатись з антигенпрезентуючою клітиною. Щоб утворився так званий імунологічний синапс між клітинами, недостатньо лише TCR і МНС, необхідні інші корецептори. У Т-лімфоциті таким є CD28, а на APC представлений В7 рецептор. Коли відбулось з’єднання обох пар рецепторів, у Т-клітині запускається каскад внутрішньоклітинних реакцій, що активують необхідні для правильної роботи клітини гени. Така клітина стає активованою, дієздатною. Після активації циркулюючі наївні Т-лімфоцити можуть мати три основні шляхи існування: 1) популяція Т-клітин може зменшуватись через реалізацію імунної відповіді шляхом апоптозу; 2) імунні клітини можуть демонструвати виснажений фенотип через повторну стимуляцію антигенами в низьких дозах і з низькою спорідненістю, наприклад при хронічній інфекції; 3) частина ефекторних клітин бере участь у довгостроковій імунній пам’яті. Т-клітини пам’яті налаштовані на більш енергійну реакцію на один і той самий антиген під час наступних зустрічей з ним, що робить їх важливими медіаторами імунної відповіді на патогени і пухлини [10].
Блокатори імунних контрольних точок
У мікрооточенні пухлин відбувається велика кількість комплексних взаємодій між пухлинними клітинами, імунними клітинами (APC, Т-клітини, NК, В-клітини тощо) і пухлинною стромою [12] (рис. 2).
Імунні контрольні точки — це важливі імунні регулятори в підтриманні імунного гомеостазу і попередженні автоімунітету. Вони включають обидва види сигналізації, інгібіторну і стимуляторну, що важливі для підтримання толерантності до власних клітин, а також для регулювання типу, вираженості і тривалості імунної відповіді. У нормальних умовах імунні контрольні точки дозволяють імунній системі відповідати на інфекції і малігнізацію клітин, захищаючи здорові тканини від будь-якого пошкодження, що може впливати на їх функцію [13].
Однак експресія деяких з цих контрольних точок пухлинними клітинами порушує регуляцію протипухлинного імунітету і посилює ріст та поширення пухлинних клітин.
Рис. 3 узагальнює ці імунні контрольні точки і їх мішені. Протипухлинна терапія, направлена на імунні контрольні точки, має на меті дію на регуляторні шляхи таким чином, щоб забезпечити імунну реакцію на пухлинні клітини. Зараз найбільш широко вивченими імунними контрольними точками є інгібуючі шляхи, що включають глікопротеїн цитотоксичних Т-лімфоцитів 4 (CTLA-4), рецептор запрограмованої клітинної смерті 1 (PD-1) та ліганд запрограмованої клітинної смерті 1 (PD-L1) [10].
Блокатори рецепторів CTLA-4
Як зазначалося вище, існує підклас Т-лімфоцитів, що пом’якшують імунну відповідь, — Т-регуляторні клітини. Вони на своїй поверхні містять рецептор CTLA-4 з високою афінністю й авідністю до В7 рецептора APC. Цей рецептор — прямий конкурент CD28 рецептора, і він запобігає активації Т-лімфоцитів. Також CTLA-4 рецептори можуть експресуватися на Т-лімфоцитах вже після активації, більшою мірою при перебуванні в лімфоїдній тканині. У периферичних тканинах після активації на Т-лімфоцитах з’являються рецептори PD-1 (рис. 4).
/47_2.jpg)
Пригнічення імунної відповіді за допомогою рецептора CTLA-4 може відбуватися на двох етапах. Перший — якщо перед активацією Т-лімфоцита він не має змоги своїм рецептором CD28 приєднатися до рецептора В7 APC, бо останній більш сильно зв’язаний з CTLA-4 Т-регуляторного лімфоцита. Таким чином, не відбувається активація Т-лімфоцита і він не виконує свою подальшу функцію. Другий етап — коли активований Т-лімфоцит намагається розпізнати чужорідний антиген на APC для подальшого розпізнавання його в організмі і утилізації. Оскільки він вже активований, то на поверхні клітини вже наявні рецептори CTLA-4. Вони конкурують з CD28 і при зв’язуванні клітини з CTLA-4 замість CD28 Т-лімфоцит стає анергічним, тобто не здатним у подальшому виконувати свою функцію [10].
Команда науковців під керівництвом Джеймса Еллісона розробила перше моноклональне антитіло, яке здатне зв’язуватись з рецептором CTLA-4. Це перешкоджає зриву активації Т-лімфоцита і процесу антигенної презентації. Вони довели, що блокада рецепторів CTLА-4 індукує довготривалу імунологічну пам’ять [14]. У випадку злоякісних пухлин блокатори рецепторів CTLА-4 посилюють клональну відповідь Т-лімфоцитів на пухлиноасоційовані неоантигени, а посилене неоантигенне навантаження має розглядатися як предиктор ефективності даного лікування. Проте підвищене пухлинне навантаження корелює зі зниженою відповіддю на лікування анти-CTLА-4, оскільки більші за розміром пухлини сприяють формуванню більш надійного протизапального пухлинного мікрооточення (рис. 5).
/47.jpg)
Застосування іпілімумабу (людське анти-CTLА-4 моноклональне антитіло) схвалено FDA в 2011 році для лікування хворих на меланому І–ІV стадії. На жаль, результати досліджень із застосування іпілімумабу у хворих на рак нирки, дрібноклітинний та недрібноклітинний рак легені та рак передміхурової залози виявили значно меншу ефективність препарату, ніж у хворих на меланому [10].
Загалом поточні дані свідчать про те, що найбільш важливим фактором прогнозу успіху лікування є співвідношення ефекторних та регуляторних Т-лімфоцитів, що інфільтрують пухлину [15].
Блокатори рецепторів PD-1/PD-L1
Рецептор PD-1 вперше був визначений у 1992 році як передбачуваний медіатор апоптозу Т-лімфоцитів, хоча пізніші дані свідчать про його роль у стримуванні гіперактивації імунної системи, аналогічної CTLA4. Експресія PD-1 на поверхні Т-клітин відбувається внаслідок стимуляції TCR, і він має здатність зв’язуватись з гомологічними до рецептора B7 антигенпрезентуючої клітини рецепторами PD-L1 та PD-L2, які конститутивно присутні на APC і можуть з’являтися у негемопоетичних тканинах під дією прозапальних цитокінів. Рецептор PD-1 має тенденцію до гальмування локальної активації Т-лімфоцитів у периферичних тканинах. Загалом PD-1 відіграє унікальну роль у підтриманні толерантності Т-лімфоцитів до клітин власного організму. Він стримує імунні реакції, головним чином шляхом внутрішньоклітинної передачі інгібуючих сигналів в ефекторних та регуляторних Т-клітинах [16].
На відміну від CTLA-4 вісь PD-1 є важливою для контролю тривалої активації та проліферації диференційованих ефекторних Т-лімфоцитів. Рецептор PD-1, взаємодіючи зі своїми лігандами, може індукувати стан дисфункції Т-клітин, який називається виснаженням Т-клітин. Але наразі залишається незрозумілим, у яких ситуаціях PD-1 опосередковує апоптоз, а в яких виснаження клітини. Окрім регуляції звичайних клітин, PD-L1 — ліганд PD-1, що знаходиться на APC, може контролювати диференціювання Т-регуляторних клітин і їх супресивну активність. Пухлинні клітини можуть використовувати механізм регуляції PD-L1 для індукції виснаження Т-клітин і створення мікросередовища пухлини, що сприяє її росту, інвазії та поширенню [16].
Активовані Т-клітини експресують рецептор PD-1, який взаємодіє зі своїм специфічним лігандом (PD-L1 або PD-L2), для пригнічення активації. Блокування осі PD-1 шляхом введення анти-PD-1 (або анти-PD-L1, або анти-PD-L2) антитіла запобігає цій інгібуючій взаємодії і обумовлює протипухлинну активність Т-лімфоцитів, сприяючи підвищеній активації і проліферації Т-клітин, посилюючи їх ефекторні функції та підтримуючи формування клітин пам’яті (рис. 6). Таким чином, більша кількість Т-лімфоцитів через TCR зв’язується з пухлинними антигенами, які представлені на клітинах пухлин молекулами MHC. Це в кінцевому підсумку призводить до вивільнення цитолітичних медіаторів, таких як перфорин та гранзим, викликаючи посилене знищення пухлини [10]. Надлишкова експресія PD-L1 клітинами пухлини пов’язана з більш успішним лікуванням препаратами, що блокують вісь PD-1. Проте численні дослідження показали негативну кореляцію між іншими білками, що беруть участь в осі PD-1, та прогнозом, що вказує на їх низьку придатність як потенційних біомаркерів [17].
/48.jpg)
У 2014 р. FDA схвалило використання лікарських засобів на основі гуманізованих і повністю людських анти-PD-1 моноклональних антитіл пембролізумаб і ніволюмаб, відповідно для лікування хворих з рефрактерною та/або нерезектабельною меланомою. У 2015 році пембролізумаб був схвалений для лікування недрібноклітинного раку легені. Додаткові успішні клінічні випробування розширили використання пембролізумабу для плоскоклітинної карциноми голови та шиї, лімфоми Ходжкіна, уротеліальної карциноми, раку шлунка та пухлин будь-якої локалізації з наявним позитивним біомаркером високої мікросателітної нестабільності (MSI-H) [15]. Після схвалення пембролізумабу для лікування пухлин будь-якої локалізації з наявним позитивним біомаркером MSI-H він став першим в історії протипухлинним препаратом, який був затверджений для лікування пухлин на основі експресії молекулярного біомаркера, а не морфологічного типу тканини, з якої походить пухлина. Однак імунодепресивний вплив мікросередовища різних пухлин ускладнює прогнозування того, для яких пацієнтів дане лікування виявиться ефективним [18].
Аналізи довгострокової виживаності хворих демонструють тривалий імуноопосередкований протипухлинний ефект блокади PD-1 та CTLA-4. Блокада PD-1 продемонструвала більшу клінічну ефективність, ніж лікування анти-CTLA-4, але причина такої розбіжності залишається незрозумілою. Існує гіпотеза, що різниця може полягати в тому, що вісь PD-1 часто взаємодіє з клітинами пухлини через експресію ліганда, тоді як CTLA-4 являє собою більш широкий імунорегуляторний ланцюг.
PD-L1 також можна блокувати за допомогою специфічних антитіл, які довели свою ефективність у лікуванні багатьох злоякісних пухлин. У 2016 році перше орієнтоване на PD-L1 гуманізоване моноклональне антитіло атезолізумаб було затверджено для лікування уротеліальної карциноми. З тих пір показання розширились, включаючи лікування дрібноклітинного та недрібноклітинного раку легені і тричі негативного раку молочної залози. Додаткові людські моноклональні антитіла до PD-L1, авелумаб і дурвалумаб, надійшли на ринок у 2017 році. Авелумаб використовується для лікування карциноми Меркеля, уротеліальної карциноми та поширеної нирковоклітинної карциноми. Дурвалумаб застосовується при уротеліальній карциномі та недрібноклітинному раку легені. Отже, подібно до PD-1, блокада PD-L1 є ефективною і відкриває нові перспективи у лікуванні злоякісних пухлин різної локалізації [19].
Інші блокатори імунних контрольних точок
Існують інші блокуючі і стимулюючі шляхи, що можуть бути використані як мішені для терапії блокаторами імунних контрольних точок. Такі лікарські засоби зараз перебувають на І/ІІ фазі клінічних випробувань.
Серед блокуючих шляхів є асоційовані з Т-клітинами інгібіторні молекули: лімфоцитактивуючий ген 3 (LAG-3, CD223), Т-клітинний імуноглобулін-3 (TIM-3), Т-клітинний імуноглобулін і ІТІМ домен (TIGIT), V-домен супресорного імуноглобуліну Т-клітинної активації (VISTA), В7-гомолог-3 (B7-H3, CD276), рецептор аденозину А2 (A2aR) і CD73, B- і T-лімфоцитарний атенюатор (BTLA, CD272). А також шляхи, не пов’язані з Т-клітинними інгібіторними молекулами: трансформуючий фактор росту β (TGF-β), кілерний імуноглобулінподібний рецептор (KIRs, CD158), фосфоінозитид 3-кіназа гамма (PI3Kγ), CD47 [20].
На противагу попереднім існують стимулюючі шляхи, які можуть мати потенційний ефект у перебудові імунної відповіді на боротьбу з пухлиною. Клітини злоякісних пухлин у процесі канцерогенезу інгібують ці шляхи. Серед них OX40 (CD134), глюкокортикоїдіндукований рецептор фактора некрозу пухлини (GITR), індуцибельний Т-клітинний костимулятор (ICOS), індуцибельний костимуляторний рецептор (4-1BB, CD137), CD27-CD70, CD40. У літературі фігурують також інші потенційні шляхи впливу на перепрограмування імунної відповіді. Зокрема, це можна зробити через індолеамін-2,3-діоксигеназу (IDO), Toll-подібні рецептори (TLRs), рецептори до IL-2, інгібітори аргінази, IL-10, онколітичі пептиди (LTX-315) [20].
Побічні ефекти при терапії блокаторами імунних контрольних точок
Блокування центральних імунних контрольних точок, що природно наявні в організмі і беруть участь в імунній регуляції, може призводити до порушення толерантності до власних тканин [21]. На підставі метааналізу даних клінічних досліджень побічні ефекти, пов’язані з імунною системою, спостерігались у 15–90 % пацієнтів. Більш тяжкі події, що вимагають втручання та/або відміни лікування, спостерігаються у 30 та 15 % пацієнтів, які отримували інгібітори CTLA-4 та PD-1 відповідно [22]. Загальною особливістю імунної токсичності терапії блокаторами імунних контрольних точок є втрата наявних Т-лімфоцитів та накопичення надмірно активних Т-клітин пам’яті, які вражають шлунково-кишковий тракт та легені, спричинюючи там запальне ураження [23]. Терапія анти-CTLA-4 несе підвищений ризик серйозних автоімунних ускладнень порівняно з терапією, спрямованою на вісь PD-1. Крім того, дані досліджень щодо підвищення дози доводять твердження, що анти-CTLA-4 препарати викликають дозозалежні відповіді, яких не спостерігається при терапії, спрямованій на вісь PD-1 [24].
Токсичні ефекти, що впливають на шлунково-кишковий тракт і головний мозок, частіше зустрічаються при терапії анти-CTLA-4, тоді як пацієнти, які отримують терапію PD-1, мають підвищений ризик гіпотиреозу, гепатотоксичності та пневмоніту. Однак із збільшенням кількості показань до лікування блокаторами контрольних точок і збільшенням кількості пролікованих пацієнтів виявлялись рідкісні побічні ефекти та неоднорідні реакції у разних органах [23]. Наприклад, гіперпрогресія захворювання спостерігалась у меншої кількості пацієнтів з різними типами пухлин, які отримували інгібітор PD-1. Зовсім недавно було показано, що інгібітор PD-1 ніволумаб може призвести до швидкого прогресування захворювання у дорослих пацієнтів з Т-клітинним лейкозом/лімфомою (ATL). Це свідчить про роль Т-регуляторних клітин, що інфільтрують пухлину, у патогенезі цієї лімфоми [25]. Для кращої класифікації реакцій пацієнтів на терапію блокаторами імунних контрольних точок було розроблено кілька критеріїв імунної відповіді. Крім того, ці критерії мають на меті відрізнити прогресію від псевдопрогресії — явища, при якому у пацієнтів, які отримують лікування інгібіторами CTLA-4 або PD-1, спостерігається період прогресування, що супроводжується подальшою швидкою регресією пухлини. Загалом блокада контрольних точок призводить до автоімунної токсичності із специфічним для терапії залученням органів [26].
Клінічне лікування токсичності, пов’язаної з терапією, однакове при застосуванні всіх блокаторів імунних контрольних точок. Токсичність оцінюється за шкалою тяжкості відповідно до 2009 National Cancer Institute Common Terminology Criteria for Adverse Events. Помірна токсичність (ступінь 1) зазвичай не лікується. При встановленні побічних явищ 2-го або 3-го ступеня терапія інгібіторами контрольних точок припиняється, доки симптоми та лабораторні показники не нормалізуються. Глюкокортикоїди також використовуються для ефективного контролю імунної гіперактивності. Інфліксимаб та інші імунодепресанти можуть застосовуватися у разі неефективності глюкокортикоїдів. Токсичність, що загрожує життю (ступінь 4), вимагає повного припинення терапії та інтенсивної терапії по життєвим показанням. Рекомендується активний моніторинг симптомів та лабораторних показників з метою запобігання смерті внаслідок блокування контрольних точок (ступінь 5) [23].
Перспективи подальшої розробки блокаторів імунних контрольних точок
Нещодавні дослідження мали на меті модифікувати існуючі антитіла і розробити нові методи їх доставки для поліпшення профілю побічних ефектів та клінічної відповіді на препарати. Наприклад, було показано, що аномальна переробка CTLA-4 та подальша деградація лізосом є механізмом, який сприяє токсичності та зниженню ефективності лікарських засобів [27]. Додаткові дослідження були зосереджені на розробці біоматеріалів для локального введення інгібіторів контрольних точок. Наприклад, у порівнянні із системною доставкою трансдермальна доставка анти-PD-1 антитіл через пластир переносилась краще і призвела до більш стійкої протипухлинної реакції на меланому у моделі на мишах. Широка галузь досліджень у даний час спрямована на відкриття нових методів зменшення токсичності, пов’язаної з терапією контрольних точок, та покращення клінічного результату при пухлинах різної локалізації [28]. Поточні дослідження спрямовані на виявлення прогностичних біомаркерів щодо органоспецифічної токсичності внаслідок терапії блокаторами контрольних точок. Наприклад, активація нейтрофілів корелює з гастроінтестинальними побічними ефектами у пацієнтів при застосуванні іпілімумабу. Збільшення кількості еозинофілів та вивільнення прозапального цитокіну IL-17 пов’язано з токсичністю незалежно від ураженого органа. Фармакогеномічне профілювання (використання генетичної інформації для прогнозування реакції на ліки) може забезпечити більше розуміння відповідності генів та шляхів опосередкування токсичності. Зрештою, є сподівання, що генетичне, біохімічне або метаболічне профілювання може бути або попереднім скринінгом, або швидко виявляти пацієнтів, які можуть мати тяжкі побічні реакції на терапію блокаторами контрольних точок [23].
Комбінована терапія блокаторами імунних контрольних точок
Після клінічного успіху монотерапії блокаторами імунних контрольних точок комбінована терапія, яка поєднує препарати з різними механізмами дії, збільшила успіх лікування різних злоякісних пухлин. Наприклад, комбінована терапія іпілімумабом та ніволумабом збільшувала показники виживаності хворих на метастатичну меланому та нирковоклітинний рак, що дозволило FDA схвалити застосування даного лікування. Синергізм дії терапії проти CTLA-4 та PD-1 обумовлений співдружньою регуляцією протипухлинного імунітету. Перехрестя між шляхами CTLA-4 та PD-1, опосередковане димеризацією CD80 та PD-L1, надає додаткове розуміння механізмів успішного застосування подвійної терапії. Однак, як і слід було очікувати, комбінована терапія блокаторами імунних контрольних точок також збільшує ризик виникнення медикаментозної токсичності.
Застосування блокаторів імунних контрольних точок з променевою терапією може бути ефективним варіантом протипухлинного лікування. Імуномодулюючий ефект лише радіотерапії має як позитивні, так і негативні сторони. Механічно променева терапія збільшує різноманітність протипухлинних Т-клітинних реакцій шляхом опромінення нових неоантигенів одночасно з пригніченням імунної відповіді шляхом індукції експресії PD-L1 на пухлинних клітинах. Отже, на основі доклінічних даних, поєднання радіотерапії з блокаторами осі PD-1 представляє собою привабливу комбінацію протипухлинного лікування. Хворі на метастатичні форми злоякісних пухлин можуть представляти цільову популяцію для застосування цієї комбінації, оскільки абскопальна реакція на променеву терапію посилюється блокуванням контрольних точок для багатьох типів пухлин. Загалом подвійна терапія інгібіторами імунних контрольних точок та комбінація променевої терапії і блокатора імунної контрольної точки представляють перспективні шляхи для синергетичних терапевтичних реакцій, оскільки ці комбінації демонструють унікальну та взаємодоповнюючу фармакодинаміку [29].
Адаптивна Т-клітинна терапія
Адаптивна Т-клітинна терапія (АTКТ), при якій автологічні або алогенні Т-клітини вводять хворим із злоякісними пухлинами, показала значну перспективу в останні роки. Життєздатність цього виду терапії вперше була показана К.М. Саутамом та співавт. у 1966 р., коли у половини пацієнтів із неоперабельними злоякісними пухлинами спостерігалася регресія пухлини після одночасної автологічної трансплантації лейкоцитів та клітин пухлини пацієнта [30]. Алогенна трансплантація гемопоетичних стовбурових клітин хворим на лейкемію являла собою перший ефективний підхід, що застосувався в клінічній практиці і продемонстрував клінічне поліпшення в пацієнтів через відповідь трансплантата Т-клітин проти пухлинних клітин [31].
АТКТ з пухлиноінфільтруючими лімфоцитами
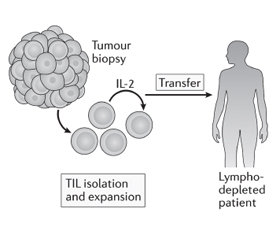
Пухлиноінфільтруючі лімфоцити (TIL) виділяють з біоптату пухлини пацієнта та культивують ex vivo за допомогою інтерлейкіну-2. Після чого їх вводять пацієнту, який переніс лімфодеплецію (виснаження лімфатичної системи) [32], щоб забезпечити для введених TIL здатність до розмноження, діяльності як ефекторних клітин і генерування імунологічної пам’яті. Оскільки Т-клітини були отримані з пухлини, передбачається, що значна частина може розпізнавати асоційовані з пухлиною антигени або неоантигени [10].
АТКТ з використанням TIL вперше стала застосовуватись для лікування хворих на меланому у 1980-х роках. Лімфоцити, виділені з біоптату пухлини, розмножувались за допомогою IL-2, а потім вводились внутрішньовенно болюсно тому самому пацієнту разом з IL-2. Коефіцієнт об’єктивної відповіді на терапію становив 34 %, однак середня тривалість клінічного ефекту становила лише 4 місяці, і лише у небагатьох пацієнтів спостерігалася повна регресія. Пізніші дослідження, що включали лімфодеплецію (за допомогою хіміотерапії або опромінення) до терапії АТКТ, у 93 пацієнтів з метастатичною меланомою були більш успішними, з повною регресією пухлини у 20 (22 %) пацієнтів, у 19 з яких все ще спостерігалась повна ремісія через 3 роки після лікування [33]. Скринінг та збагачення неоантигенспецифічних TIL, що стало можливим завдяки високопродуктивним технологіям, нещодавно продемонстрував ефективність у пацієнток з метастатичним раком молочних залоз. Однак для того, щоб АТКТ на основі TIL викликала довготривалі реакції (рис. 7), необхідна наявність в пухлині ефекторних Т-лімфоцитів з протипухлинною активністю, що спостерігається рідко. Інші інноваційні підходи до налаштування активності та проліферації Т-лімфоцитів можуть дозволити розробити більш широкий спектр методів АТКТ [10].
/50.jpg)
Сконструйовані лімфоцити для адаптивної Т-клітинної терапії
Проблеми, пов’язані з розмноженням специфічних для пухлини Т-лімфоцитів in vitro, призвели до спроб створення лімфоцитів, сконструйованих за допомогою T-клітинного рецептора. Фізіологічний Т-клітинний рецепторний (TCR) комплекс набуває своєї специфічності завдяки поліморфним ланцюгам α- та β-глікопротеїнів, які мають антигензв’язувальну частину, а також має консервативний (постійний) домен, що представлений групою неполіморфних білків, CD3 γ, δ, ε та ζ. Біоінженерія α- та β-глікопротеїнового антигензв’язуючого домену, зберігаючи консервативні домени (Cα та Cβ), дозволяє розвивати та розширювати Т-лімфоцити, що реагують специфічно на пухлинні неоантигени [10] (рис. 8).
Однак ці клітини обмежуються реакцією на пухлинні антигени, представлені МНС, а не на поверхневі антигени на клітинах пухлини. Тому були винайдені синтетичні химерні антигенні рецептори (chimeric antigen receptor, CAR), які можуть обходити обмеження МНС та направляти специфічну цитотоксичність до молекули-мішені на поверхні клітини злоякісної пухлини (рис. 9) [34]. Ізольовані від пацієнта (або алогенного донора) Т-лімфоцити генетично модифікуються для експресії CAR, а потім розмножуються та вводяться пацієнту. Це дозволяє подолати проблему, пов’язану з тим, що клітини пухлини часто знижують регуляцію молекул МНС, що залишає клітину нездатною «показувати» антиген звичайним Т-клітинам. CAR складаються з антигензв’язуючого домену, найчастіше із варіабельних ділянок антитіл, пов’язаних із сигнальними доменами TCR та різними костимулюючими молекулами.
/51.jpg)
Спочатку химерні антигенні рецептори складались із позаклітинного одноланцюгового фрагмента варіабельної ділянки антитіла, з’єднаного з CD3 ζ-сигнальним доменом. Низька проліферативна та функціональна активність цих CAR першого покоління призвела до розвитку CAR другого і третього покоління, що містять внутрішньоклітинні модулі з костимулюючих молекул (CD28 та/або 4-1BB), що забезпечують додаткові сигнали, необхідні для повної активації Т-клітини. Наступні покоління CAR Т-клітин містять подальші модифікації для поліпшення протипухлинної ефективності. Наприклад, «броньовані» CAR Т-клітини четвертого покоління розроблені для секреції прозапальних цитокінів, таких як IL-12, необхідних для подолання імуносупресії в мікросередовищі пухлини. Химерний рецептор цитокінів 4αβ, що включає ектодомен IL-4Rα, злитий з ланцюгом IL-2/IL-15Rβ, сигналізує у відповідь на IL-4, який часто представлений у багатьох типах пухлин [10]. CAR Т-клітин зазвичай розробляються з використанням ретровірусної трансдукції, проте в останніх дослідженнях використовували технологію CRISPR — Cas9. Технологія CRISPR — Cas9 може бути використана для безпосереднього редагування послідовності зародкових ліній TCR, що може призвести до більш рівномірного генерування CAR Т-клітинами і, зрештою, до більшої ефективності [35]. Обмеженнями до розвитку терапії CAR Т-клітин є вимога до чіткого тканинного антигену, обмеженого і специфічного лише для клітин пухлини. Наприклад, CAR Т-клітини, сконструйовані зі специфічністю для поверхневої молекули CD19, яка експресується усіма В-клітинами, ефективні для лікування злоякісних пухлин з В-клітин. Перше клінічне випробовування CD-специфічних CAR Т-клітин другого покоління призвело до стійкої ремісії при хронічному лімфолейкозі. Додаткові клінічні випробування CD19-специфічних CAR Т-клітин другого покоління при В-клітинному гострому лімфобластному лейкозі (B-ГЛЛ) зафіксували ремісію у всіх пацієнтів з B-ГЛЛ, які брали участь у дослідженні. Подальші спостереження за пацієнтами з B-ГЛЛ, які брали участь у цьому клінічному дослідженні, зафіксували повну ремісію захворювання у 44 із 53 (83 %) пацієнтів при медіані спостереження 29 місяців [36, 37]. Подібні успіхи були зареєстровані у пацієнтів з дифузною В-великоклітинною лімфомою, що призвело до схвалення FDA у 2017 році такого методу лікування хворих на В-клітинні злоякісні пухлини. Перехресне таргетування нормальних CD19+ В-клітин не перешкоджає терапії та не викликає серйозних побічних ефектів. Однак, навіть за такої ідеальної мішені, втрата пухлинними клітинами антигену CD19 є частою причиною неефективності лікування.
CD22 — ще один антиген, який зазвичай експресується клітинами пухлини при B-ГЛЛ і показав перспективність як мішень для терапії CAR Т-клітинами в фазі I клінічного дослідження [38]. Гемобластози, які не експресують CD19, а також солідні пухлини у даний час досліджуються на інші мішені, особливо пухлинні неоантигени. Терапія CAR T-клітинами, спрямована на антиген дозрівання В-клітин (BCMA), була готова до затвердження FDA для лікування множинної мієломи в 2020 році на основі даних, отриманих у доклінічних та клінічних випробуваннях. Однак дослідження додаткових антигенів-мішеней триває через повідомлення про рецидиви захворювання. У нещодавніх доклінічних дослідженнях виявлено ще один цільовий антиген, GPRC5D, з порівнянною ефективністю та токсичністю до терапії CAR Т-клітинами, спрямованої на BCMA. Зараз терапія мала лише помірний успіх при солідних пухлинах, і зараз вводяться інноваційні підходи до вдосконалення терапії. Нещодавно визначена мішень B7-H3 (також відомий як CD276) для даної терапії продемонструвала успіх у багатьох моделях дитячих солідних пухлин [39].
Ефективність CAR Т-клітинної терапії може бути посилена за допомогою спільної експресії химерного рецептора цитокінів (4αβ), який стимулює проліферацію у відповідь на IL-4, цитокін, якого зазвичай багато в мікросередовищі пухлини. Попередні дослідження показали, що такий підхід працює для CAR Т-клітин, спрямованих проти різних асоційованих з пухлиною антигенів, і зараз проводяться клінічні випробування ефективності застосування у хворих з пухлинами голови та шиї. Крім того, було показано, що надмірна експресія фактора транскрипції JUN надає стійкість до виснаження CAR Т-клітин [40].
Побічні ефекти та обмеження при терапії адаптивними Т-клітинами
Унаслідок терапії CAR Т-клітинами може виникати токсичність, що впливає на різні системи органів із різним ступенем тяжкості. Пацієнти найчастіше відчувають синдром вивільнення цитокінів (CRS, за клінікою схожий на цитокіновий шторм, але принципово відрізняється від нього) та нейротоксичність. CRS є результатом потужної активації та проліферації CAR T-клітин in vivo і зазвичай з’являється швидко після перенесення клітин в організм пацієнта. Симптоми цього ускладнення часто бувають легкими, грипоподібними, але можуть й загрожувати життю пацієнта, включаючи гіпотонію, гіпертермію (лихоманку), перерозподіл рідини в організмі, коагулопатію та поліорганну недостатність. Також можуть траплятися серйозні неврологічні ускладнення, такі як пов’язана з CAR Т-клітинною терапією енцефалопатія, яка, як правило, характеризується сплутаністю свідомості та маренням, але іноді також супроводжується судомами та набряком мозку [41]. Глюкокортикоїди є першою лінією лікування легких форм CRS та енцефалопатії, пов’язаної з CAR Т-клітинною терапією. Тоцилізумаб, рекомбінантне гуманізоване моноклональне антитіло проти рецептора IL-6, є високоефективним препаратом другої лінії лікування CRS, спричиненого терапією CAR Т-клітинами [42]. До інших побічних ефектів CD19-специфічної терапії CAR T-клітинами відносять лімфопенію та гіпогаммаглобулінемію, які можна ефективно контролювати внутрішньовенною терапією імуноглобулінами, подібно до лікування, яке отримують пацієнти з первинними В-клітинними імунодефіцитами. Механізми цих побічних ефектів поки незрозумілі, і подальші дослідження можуть допомогти знайти способи уникнути токсичності або мінімізувати її [43].
Застосування ATКТ вимагає розробки специфічної індивідуальної для пацієнта терапії, а виробництво препаратів високотехнологічне, тому її вартість може бути надмірною, що обмежує доступ пацієнта до лікування. У Сполучених Штатах за 2020 рік вартість терапії CAR Т-клітинами становила від 373 000 до 475 000 доларів США на пацієнта. Однак ці суми не враховують додаткові витрати, пов’язані з лікуванням серйозних побічних ефектів, загальних для CAR Т-клітинної терапії, які, за оцінками, збільшують витрати, пов’язані з ліками, на 30 000 доларів США або більше. Порівняно з терапією CAR Т-клітинами блокатори імунних контрольних точок мають вартість близько 12 500 доларів США на місяць. Доступність терапії CAR Т-клітинами також є серйозною проблемою для пацієнтів, оскільки існує лише декілька лабораторій, сертифікованих для генерування CAR Т-клітин, і лише кілька спеціалізованих центрів третинної допомоги в США та Європі (Німеччина, Італія), здатних проводити цю терапію. Нарешті, мінливість у виробництві CAR Т-клітин та відсутність стандартизації можуть сприяти неоднаковим результатам лікування [42, 44].
Висновки
Успіх у лікуванні хворих на злоякісні пухлини імунотерапевтичними протипухлинними засобами підкреслює важливість розуміння імунології пухлин, особливо ролі пухлинних антигенів та імуносупресивного впливу мікросередовища пухлини. З 1980-х років було розроблено багато нових ефективних засобів імунотерапії злоякісних пухлин. Однак, незважаючи на ці досягнення, необхідні подальші дослідження для створення нових та підвищення ефективності існуючих лікарських засобів. Багато підходів на основі цитокінів, численні моноклональні антитіла та їх похідні стали стандартним методом лікування різних злоякісних пухлин, інші ж підходи до імунотерапії, наприклад адаптивна клітинна терапія, залишаються більшою мірою експериментальними. Імунотерапія злоякісних пухлин продовжує розвиватися, оскільки розширюються показання до затверджених у даний час методів лікування та продовжуються пошуки нових антигенів клітин пухлини, що слугують мішенями для лікарських засобів. Сьогодні багато нових стратегій та засобів імунотерапії досліджуються та перевіряються в клінічних випробуваннях, і, можливо, вони забезпечать нові ефективні методи лікування для пацієнтів зі злоякісними пухлинами.
Конфлікт інтересів. Авторам невідомі будь-які приналежності, членство чи фінансові вкладення, які можуть сприйматися як такі, що впливають на об’єктивність цього огляду.
Отримано/Received 19.07.2021
Рецензовано/Revised 27.07.2021
Прийнято до друку/Accepted 05.08.2021

/43.jpg)
/45.jpg)
/46.jpg)
/47_2.jpg)
/48.jpg)
/51.jpg)