
Журнал «Здоровье ребенка» 6 (66) 2015
Вернуться к номеру
Синдром Ангельмана Часть 2 (клиника и диагностика)
Авторы: Абатуров А.Е., Петренко Л.Л., Кривуша Е.Л. - ГУ «Днепропетровская медицинская академия Министерства здравоохранения Украины»
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
В статье представлены основные клинические проявления синдрома Ангельмана (AS). Статья содержит современные данные об особенностях физического, интеллектуального, речевого и полового развития больных с AS. Представлены данные о взаимосвязи клинических особенностей AS с характером генетических нарушений. В статье приведены клинические критерии диагностики синдрома Ангельмана с учетом частоты встречаемости основных клинических признаков. Авторами рассмотрено целевое назначение молекулярно-генетических анализов, используемых для верификации диагноза и определения генетического механизма AS. Статья содержит алгоритм лабораторно-диагностических действий проведения молекулярно-генетического обследования пациентов с AS.
У статті представлені основні клінічні прояви синдрому Ангельмана (AS). Стаття містить сучасні дані про особливості фізичного, інтелектуального, мовного та статевого розвитку хворих з AS. Представлені дані про взаємозв’язок клінічних особливостей AS із характером генетичних порушень. У статті надані клінічні критерії діагностики синдрому Ангельмана з урахуванням частоти зустрічаємості основних клінічних ознак. Авторами розглянуто цільове призначення молекулярно-генетичних аналізів, використовуваних для верифікації діагнозу та визначення генетичного механізму AS. Стаття містить алгоритм лабораторно-діагностичних дій проведення молекулярно-генетичного обстеження пацієнтів з AS.
The article presents the main clinical manifestations of Angelman syndrome. The article contains current data about the features of the physical, intellectual, verbal and sexual development of patients with Angelman syndrome. There are shown the data of the relationship of clinical features of Angelman syndrome with the nature of genetic disorders. The article presents the clinical criteria for diagnosis of Angelman syndrome in view of the incidence of key clinical signs. The authors reviewed the intended use of the molecular genetic analysis to verify the diagnosis and determine the genetic mechanism of Angelman syndrome. This article contains the algorithm of laboratory and diagnostic methods for molecular genetic exa-mination of patients with Angelman syndrome.
синдром Ангельмана.
синдром Ангельмана.
Angelman syndrome.
Статья опубликована на с. 119-125
Клинические проявления синдрома Ангельмана
Клинические проявления AS [30]
Корреляция фено- и генотипа
/121.jpg)
Критерии диагностики
/122.jpg)
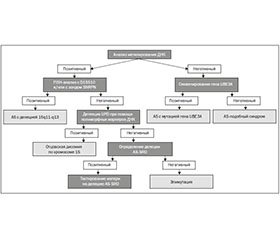
Молекулярно-генетическая диагностика
/123.jpg)
/123_2.jpg)
1. Миронов М.Б. Синдром Ангельмана. Клинический случай / М.Б. Миронов, К.Ю. Мухин, Н.Ю. Кузина и др. // Русский журнал детской неврологии. — 2009. — Т. IV, вып. 1. — С. 53-62.
2. Allen K.D. Evaluation of a behavioral treatment package to reduce sleep problems in children with Angelman Syndrome / K.D. Allen, B.R. Kuhn, K.A. DeHaai, D.P. Wallace // Res. Dev. Disabil. — 2013 Jan. — 34(1). — 676-86. — doi: 10.1016/j.ridd.2012.10.001.
3. Andersen W.H., Rasmussen R.K., Strømme P. Levels of cognitive and linguistic development in Angelman syndrome: a study of 20 children // Logoped Phoniatr. Vocol. — 2001. — 26(1). — 2-9. — PMID: 11432411.
4. Bird L.M. Angelman syndrome: review of clinical and molecular aspects //Appl. Clin. Genet. — 2014, May 16. — 7. — 93-104. — doi: 10.2147/TACG.S57386.
5. Buiting K. Clinical utility gene card for: Angelman Syndrome / K. Buiting, J. Clayton-Smith, D.J. Driscoll // Eur. J. Hum. Genet. — 2015 Feb. — 23(2). — doi: 10.1038/ejhg.2014.93.
6. Cali F. Novel deletion of the E3A ubiquitin protein ligase gene detected by multiplex ligation-dependent probe amplification in a patient with Angelman syndrome / F. Cali, A. Ragalmuto, V. Chiavetta et al. // Exp. Mol. Med. — 2010, Dec 31. — 42(12). — 842-8.
7. Clarke D.J., Marston G. Problem behaviors associated with 15q- Angelman syndrome // Am. J. Ment. Retard. — 2000 Jan. — 105(1). — 25-31. — PMID: 10683706.
8. Clayton-Smith J., Laan L. Angelman syndrome: a review of the clinical and genetic aspects // J. Med. Genet. — 2003 Feb. — 40(2). — 87-95. — doi: 10.1136/jmg.40.2.87.
9. Dagli A.I., Mueller J., Williams C.A. Angelman Syndrome // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors. GeneReviews® [Internet]. — Seattle (WA): University of Washington, Seattle; 1993-2015. — 1998, Sep 15 [updated 2015 May 14]. — PMID: 20301323.
10. Duca D.G. Diagnostic approach of Angelman syndrome / D.G. Duca, D. Craiu, M. Boer et al. // Maedica (Buchar). — 2013 Sep. — 8(4). — 321-7. — PMCID: PMC3968465.
11. Fiumara A. Epilepsy in patients with Angelman syndrome / A. Fiumara, A. Pittalà, M. Cocuzza, G. Sorge // Ital. J. Pediatr. — 2010, Apr 16. — 36. — 31. — doi: 10.1186/1824-7288-36-31.
12. Gentile J.K. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations / J.K. Gentile, W.H. Tan, L.T. Horowitz et al. // J. Dev. Behav. Pediatr. — 2010 Sep. — 31(7). — 592-601. — doi: 10.1097/DBP.0b013e3181ee408e.
13. Giroud M. Angelman syndrome: a case series assessing neurological issues in adulthood / M. Giroud, B. Daubail, N. Kha-yat et al. // Eur. Neurol. — 2015. — 73(1–2). — 119-25. — doi: 10.1159/000369454.
14. Horsler K., Oliver C. The behavioural phenotype of Angelman syndrome // J. Intellect. Disabil. Res. — 2006 Jan. — 50(Pt 1). — 33-53. — PMID: 16316429.
15. Lossie A.C. Distinct phenotypes distinguish the molecular classes of Angelman syndrome / A.C. Lossie, M.M. Whitney, D. Amidon et al. // J. Med. Genet. — 2001 Dec. — 38(12). — 834-45. — doi: 10.1136/jmg.38.12.834.
16. Lossie A.M., Driscoll D.J. Transmission of Angelman syndrome by an affected mother // Genet. Med. — 1999. — 1. — 262-266.
17. Mane S., Chatterjee R. Angelman syndrome: The blurred lines of interpretation in cognitive defects // J. Pediatr. Neurosci. — 2015 Jan — Mar. — 10(1). — 70-2. — doi: 10.4103/1817-1745.154360.
18. Margolis S.S. Angelman Syndrome / S.S. Margolis, G.L. Sell, M.A. Zbinden, L.M. Bird // Neurotherapeutics. — 2015, Jun 4. — PMID: 26040994.
19. Park S.H. Epilepsy in Korean patients with Angelman syndrome / S.H. Park, J.R. Yoon, H.D. Kim, J.S. Lee, Y.M. Lee, H.C. Kang // Korean J. Pediatr. — 2012 May. — 55(5). — 171-6. — doi: 10.3345/kjp.2012.55.5.171.
20. Peters S.U. Cognitive and adaptive behavior profiles of children with Angelman syndrome / Peters S.U., Goddard-Finegold J., Beaudet A.L., Madduri N., Turcich M., Bacino C.A. // Am. J. Med. Genet. A. — 2004. — 128A(2). — 110-113. — doi: 10.1002/ajmg.a.30065.
21. Sarkar P.A., Shigli A., Patidar C. Happy Puppet syndrome // BMJ Case Rep. — 2011, Oct 28. — 2011. — pii: bcr0920114747. doi: 10.1136/bcr.09.2011.4747.
22. Seltzer L.E., Paciorkowski A.R. Genetic disorders associated with postnatal microcephaly // Am. J. Med. Genet. C Semin. Med. Genet. — 2014 Jun. — 166C(2). — 140-55. — doi: 10.1002/ajmg.c.31400.
23. Shi S.Q. Ube3a imprinting impairs circadian robustness in Angelman syndrome models / S.Q. Shi, T.J. Bichell, R.A. Ihrie, C.H. Johnson // Curr. Biol. — 2015, Mar 2. — 25(5). — 537-45. — doi: 10. 1016/j.cub.2014.12.047.
24.Takaesu Y., Komada Y., Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients // Sleep Med. — 2012 Oct. — 13(9). — 1164-70. — doi: 10.1016/j.sleep.2012.06.015.
25. Tan W.H. Angelman syndrome: Mutations influence features in early childhood / W.H. Tan, C.A. Bacino, S.A. Skinner et al. // Am. J. Med. Genet. A. — 2011 Jan. — 155A(1). — 81-90. — doi: 10.1002/ajmg.a.33775.
26. Thibert R.L. Neurologic manifestations of Angelman syndrome / R.L. Thibert, A.M. Larson, D.T. Hsieh, A.R. Raby, E.A. Thiele // Pediatr. Neurol. — 2013 Apr. — 48(4). — 271-9. — doi: 10.1016/j.pediatrneurol.2012.09.015.
27. Valente K.D. Angelman syndrome caused by deletion: a genotype-phenotype correlation determined by breakpoint / Valente K.D., Varela M.C., Koiffmann C.P. et al. // Epilepsy Res. — 2013 Jul. — 105(1–2). — 234-9. — doi: 10.1016/j.eplepsyres.2012.12.005.
28.Varela M.C. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects / M.C. Varela, F. Kok, P.A. Otto, C.P. Koiffmann // Eur. J. Hum. Genet. — 2004 Dec. — 12(12). — 987-92. — doi:10.1038/sj.ejhg.5201264.
29. Walz N.C. Parent report of stereotyped behaviors, social interaction, and developmental disturbances in individuals with Angelman syndrome // J. Autism. Dev. Disord. — 2007 May. — 37(5). — 940-7. — doi 10.1007/s10803-006-0233-8.
30. Williams C.A. Angelman syndrome 2005: updated consensus for diagnostic criteria/ C.A. Williams, A.L. Beaudet, J. Clayton-Smith et al. // Am. J. Med. Genet. A. — 2006, Mar 1. — 140(5). — 413-8. — doi: 10.1002/ajmg.a.31074.
31. Williams C.A. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation / C.A. Williams, H. Angelman, J. Clayton-Smith et al. // Am. J. Med. Genet. — 1995, Mar 27. — 56(2). — 237-8. — PMID: 7625452.
32. Williams C.A. Neurological aspects of the Angelman syndrome // Brain Dev. — 2005 Mar. — 27(2). — 88-94. — doi: http://dx.doi.org/10.1016/j.braindev.2003.09.014.
33. Williams C.A. The behavioral phenotype of the Angelman syndrome // Am. J. Med. Genet. C Semin. Med. Ge-net. — 2010, Nov 15. — 154C(4). — 432-7. — doi: 10.1002/ajmg.c.30278.
34. Williams C.A., Driscoll D.J., Dagli A.I. Clinical and genetic aspects of Angelman syndrome // Genet. Med. — 2010 Jul. — 12(7). — 385-95. — doi: 10.1097/GIM.0b013e3181def138.