
Международный неврологический журнал 1 (71) 2015
Вернуться к номеру
Оптикомиелит (болезнь Девика). Научный обзор и собственное клиническое наблюдение
Авторы: Мироненко Т.В., Хубетова И.В. - ГУ «Луганский государственный медицинский университет»
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
Авторами проведен анализ данных отечественной и иностранной литературы по изучаемой проблеме. Обобщены взгляды на проблему этиопатогенеза, классификацию, клинико-диагностический алгоритм оптикомиелита Девика.
Проведена дифференциация оптикомиелита и рассеянного склероза, острого демиелинизирующего энцефаломиелита, поперечного некротизирующего миелита и др. Представлены общие схемы лечения описываемого синдрома.
В качестве иллюстрации использовано собственное клиническое наблюдение, подтвержденное результатами нейровизуализации. Обоснована практическая направленность изложенного научного сообщения.
Авторами проведено аналіз даних вітчизняної та іноземної літератури з досліджуваної проблеми. Узагальнено погляди на проблему етіопатогенезу, класифікацію, клініко-діагностичний алгоритм оптикомієліту Девіка.
Проведена диференціація оптикомієліту з розсіяним склерозом, гострим демієлінізуючим енцефаломієлітом, поперечним некротизуючим мієлітом. Представлені загальні схеми лікування синдрому, що описується.
Як ілюстрацію використано власне клінічне спостереження, підтверджене результатами нейровізуалізації. Обґрунтована практична спрямованість викладеного наукового повідомлення.
The authors analyzed data of domestic and foreign li-terature on the problem under study. There were summarized views on the problem of etiopathogenesis, classification, clinical and diagnostic algorithm of Devic’s disease.
The differential diagnosis of neuromyelitics optica and multiple sclerosis, acute demyelinating encephalomyelitis, transverse necroti-zing myelitis etc. has been carried out. There are represented common treatment regimens for described syndrome.
As an illustration we used out own clinical observation confirmed by the results of neuroimaging. The practical orientation of the above scientific posts is proved.
оптиконевромиелит, клиника, диагностика.
оптиконевромієліт, клініка, діагностика.
neuromyelitics optica, clinical picture, diagnosis.
Статья опубликована на с. 141-147
Оптиконевромиелит (ОНМ, МКБ–10, G36.0) — оптикомиелит Девика, синдром или болезнь Девика — идиопатическое тяжелое воспалительное демиелинизирующее заболевание, характеризующееся избирательным вовлечением в патологический процесс зрительных нервов и спинного мозга при относительной интактности структур головного мозга.
Долгое время традиционная локализация ОНМ рассматривалась как патология центральной нервной системы в виде монофазного заболевания с двусторонним поражением зрительных нервов и одновременным или через короткий промежуток времени развитием тяжелого поперечного миелита. Однако в конце XX века определение данного заболевания стало более универсальным, допускающим существенные различия в вариантах течения (монофазный и ремиттирующий), степени вовлечения зрительных нервов (одностороннее или двустороннее поражение), последовательности развития процесса в спинном мозге и зрительных нервах, специфических особенностях течения оптического неврита и миелита (уровень поражения спинного мозга продольный и поперечный, ограниченный или распространенный, вовлечение серого или белого вещества).
Впервые случай подобного сочетания поражения зрительных нервов и спинного мозга описал С. Allbutt в 1870 г.
К 1894 г. в странах Европы и США было накоплено 16 случаев, которые вместе с собственным наблюдением E. Devic и его ученик F. Gault подробно изучили и предложили выделить в отдельную нозологическую форму [14]. Однако длительное время заболевание традиционно рассматривалось в рамках одного из злокачественных вариантов течения рассеянного склероза (РС). Прогресс в изучении патогенеза демиелинизирующих заболеваний ЦНС позволил к концу XX века считать ОНМ не редким вариантом течения РС, а отдельной нозологической формой [1, 3].
Характеризуя эпидемиологию ОНМ, следует отметить, что в европейских странах он составляет 1–2 % в структуре остальных демиелинизирующих заболеваний центральной нервной системы. Вероятно, это связано с тем, что многие случаи ОНМ нередко ошибочно трактуются как РС, рецидивирующий поперечный миелит, рецидивирующий ретробульбарный нев–рит [5].
ОНМ — заболевание аутоиммунной природы, преобладающее среди представителей неевропеоидной расы. Соотношение женщин и мужчин составляет (2–8) : 1. Возраст, в котором дебютирует заболевание, варьирует от 1 года до 77 лет, наиболее часто оно начинается в 35–47 лет [9].
В литературе описаны единичные семейные случаи оптикомиелита Девика.
Довольно часто ОНМ (50–70 %) сочетается с другими аутоиммунными заболеваниями — синдромом Шегрена, аутоиммунным тиреоидитом, ревматоидным артритом, системной красной волчанкой, пернициозной анемией, неспецифическим язвенным колитом, первичным склерозирующим холангитом, тромбоцитопенической пурпурой.
Болезнь Девика встречается в странах Юго–Восточной Азии, особенно в Японии, преобладая у женщин среднего возраста [15].
В 2004 г. впервые были выявлены специфические для ОНМ антитела NMO–IgG, которые обнаруживались у 63–73 % пациентов. Их наличие подтвердило аутоиммунные механизмы патогенеза оптиконевромиелита.
Подобные антитела отсутствуют у больных с классическим РС, при воспалительных и невоспалительных заболеваниях ЦНС, при других аутоиммунных заболеваниях и у здоровых лиц [1].
Мишенью антител является выделенный в 2005 г. аквапорин–4 — белковый канал, регулирующий водный баланс нервной клетки. Он тесно связан с гематоэнцефалическим барьером, с элементами декстрогликанового комплекса в области примыкания астроцитов к сосудистой стенке. Наибольшая концентрация аквапорина–4 в ЦНС отмечена в сером веществе спинного мозга, гипоталамусе, перивентрикулярных областях [11].
Заболевание поражает, как правило, зрительный нерв, хиазму, спинной мозг, гипоталамус, мозговой ствол. Патофизиологически при ОНМ имеют место демиелинизация, некроз белого и серого вещества. При этом воспалительный инфильтрат содержит большое количество полиморфноядерных лейкоцитов, макрофагов, эозинофилов, что отличает ОНМ от РС. Присутствует гиалиноз артерий спинного мозга среднего калибра, что обычно сопровождается некрозом спинного мозга.
В очагах демиелинизации и вокруг сосудов находят отложение компонентов комплемента и IgG, ассоциированное с фиброзом и пролиферацией сосудов [9].
Хронические очаги воспаления в мозге представлены кистозной дегенерацией, глиозом и атрофией нервной ткани, которые могут привести к развитию вторичной сирингомиелии [10].
На сегодняшний день не противоречит диагнозу ОНМ выявление очагового поражения нервной системы за пределами зрительных нервов и спинного мозга.
Описанные особенности морфологических изменений в спинном мозге могут напоминать аутоиммунное воспаление, протекающее по типу васкулита. Практически в 100 % случаев в спинальных очагах воспаления выявляется гиалинизация сосудов, поэтому присутствует компонент васкулита при ОНМ. Такие изменения не обнаруживаются в типичных очагах РС, инфарктных очагах или в очагах при остром рассеянном энцефаломиелите. В отличие от типичного РС в патогенезе ОНМ предполагается ведущая роль В–клеточно–индуцированных реакций.
Сходная иммунопатологическая реакция обсуждается при тяжелом течении ремиттирующего и вторично–прогрессирующего РС.
В ряде случаев ОНМ служит проявлением системной красной волчанки, антифосфолипидного синдрома, туберкулеза легких.
Особая роль гуморального иммунитета при ОНМ может также быть одним из основных отличий его патогенеза от РС. В пользу такого предположения свидетельствует недавнее открытие плазменных аутоантител NMO–IgG Devic [11].
Основной мишенью для иммунной реакции, как уже указывалось выше, является аквапорин–4 (AQPH). Сывороточные антитела (NMO–IgG) связываются с церебральными микрососудами, мягкой мозговой оболочкой, пространствами Вирхова — Робина. Аквапорин–4, находящийся в отростках астроцитов или покрывающий участки сосудов, не покрытых астроцитарными ножками, участвует в образовании гематоэнцефалического барьера и при его поражении не может справиться со своей функцией.
Повреждение астроцитов способствует доступу других иммунных комплексов в ЦНС и активирует формирование в ней аутоиммунного процесса [10]. При синдроме Девика поражается оболочка жироподобного вещества астроцитов, защищающая зрительные и спинномозговые нервы.
Поэтому первично в клинике заболевания наступают нарушения зрения в виде его снижения, вплоть до полной утраты, а через некоторое время присоединяются симптомы тяжелого поперечного миелита — парапарезы, тетрапарезы, нарушение функции тазовых органов.
В настоящее время допускается, что ОНМ может иметь как монофазный, так и ремиттирующий тип течения. Вместе с тем повторные атаки при нем менее типичны, как и ремиссии.
Анализируя клиническое течение ОНМ по данным 71 пациента за период 1950–1997 гг., I. Miyаzava еt al. (2002) показали, что ОНМ может быть как монофазным, так и ремиттирующим. В последнем случае в течение 3 лет развивается тяжелое обострение оптического неврита или миелита. В трети случаев причиной смерти является развитие дыхательных нарушений [12].
При общей тенденции к плохому прогнозу допускается возможность нескольких вариантов исхода. В ряде случаев предполагается полное выздоровление, другой вариант течения ОНМ — наличие ремиссий заболевания. Самый неблагоприятный вариант протекает как прогрессирующее ухудшение и смерть больного.
Анализируя прогностическую значимость типа и особенностей течения ОНМ, D. Wingerchuk и соавт. [12] отметили, что исход хуже в группе пациентов с монофазным течением заболевания, а также при стойком снижении остроты зрения и стойких параличах.
Предикторами ремиттирующей формы ОНМ являются более продолжительные интервалы между первыми двумя атаками, более старший возраст дебюта болезни, женский пол и менее тяжелый моторный дефицит при поражении спинного мозга.
Остаточный тяжелый дефицит после острой атаки чаще встречается при ОНМ, чем при РС [5], что имеет определенное дифференциальное значение.
Одним из главных симптомов ОНМ является оптический неврит. При офтальмоскопии чаще обнаруживаются нормальная картина глазного дна, небольшая стушеванность дисков зрительных нервов, легкий отек, атрофия и бледность дисков зрительных нервов в хронических случаях.
При ОНМ оптический неврит, как правило, билатеральный, обычно предшествует миелиту (в 80 % случаев). Через несколько недель, реже месяцев, развивается тяжелый поперечный миелит. В большинстве наблюдений развитие миелита происходит менее чем через 3 месяца. Однако в 20 % случаев поперечный миелит может предшествовать оптическому невриту [4].
Поражение спинного мозга при ОНМ может протекать в изолированной форме. При этом вариантом течения бывает острый частичный поперечный или продольный распространенный миелит. Развитие полного поперечного миелита при РС является достаточно редким.
Исследование, проведенное в группах больных с ОНМ, продольным распространенным миелитом, острым частичным поперечным миелитом, показало, что при наличии иммуноглобулинов NMO–IgG продольный распространенный миелит может рассматриваться как неполная форма ОНМ.
Типичными симптомами миелита служат мышечная слабость, спастичность, дискоординация, атаксия, симптом Лермитта, задержка мочи, вегетативная дисфункция, возможны сонорные расстройства ниже уровня поражения спинного мозга.
При монофазном типе течения (менее 20 % пациентов с ОНМ) одновременно развивается одно– либо двусторонний неврит зрительного нерва и поперечный миелит, затем повторные эпизоды миелита и/или оптического неврита не регистрируются.
При рецидивирующем течении ОНМ, наоборот, первые атаки оптического неврита и миелита могут быть разделены между собой во времени неделями или даже годами, но зато неврит зрительного нерва, либо миелит, либо оба эти состояния рецидивируют на протяжении ближайших месяцев или лет; на протяжении первого года рецидив возникает у 55 % пациентов, на протяжении первых 3–5 лет — соответственно у 78 и 99 % [6].
У пациентов с рецидивирующим монофазным течением болезни, несмотря на большую исходную выраженность нарушений, долгосрочный прогноз лучше, так как не происходит накопления неврологического дефицита, возникающего при повторных атаках.
В литературе имеются указания на нетипичное начало болезни Девика. Так, рядом ученых отмечено, что перед возникновением неврологических симптомов имел место продромальный период в виде лихорадки, инфекции, других аутоиммунных заболеваний.
Norajima et al. (2011) [6] сообщают об очень редком, нетипичном дебюте болезни Девика — синдроме неадекватной секреции антидиуретического гормона. По мнению авторов, при наличии указанного синдрома с отсутствием признаков поражения зрительных нервов и спинного мозга и при наличии подтвержденных МРТ очагов в гипоталамусе следует рассматривать синдром Девика как возможную причину этого состояния.
Е.Г. Менделевич и соавт. (2004) сообщают о 2 клинических наблюдениях ОНМ, осложненных сирингомиелическими полостями, сформировавшимися в местах некроза очагов воспаления в спинном мозге. Авторы высказывают предположения о патогенезе сирингомиелии в результате дисциркуляции цереброспинальной жидкости (ЦСЖ) в центральном спинномозговом канале и субарахноидальных пространствах спинного мозга при наличии зоны некроза в спинном мозге. Летальный исход ОНМ может наступить в результате тяжелой атаки миелита с вовлечением в процесс шейного отдела спинного мозга и развитием дыхательной недостаточности.
Заболевание протекает однофазно, редко бывает мультифазным. Как уже указывалось выше, оптикомиелит (синдром Девика) характеризуется односторонним, чаще одновременным двусторонним, тяжелым поражением зрительных нервов с отеком в остром периоде и атрофией впоследствии.
Изменения в зрительных нервах видны как при исследовании глазного дна, так и при нейровизуализации, последние в отличие от РС захватывают зрительные нервы на большом протяжении. Практически во всех случаях ОНМ приходится дифференцировать с РС.
В отличие от ОНМ при РС очаги демиелинизации в спинном мозге по длине обычно не превышают одного сегмента. На МРТ головного мозга при ОНМ либо не выявляется никаких патологических изменений, либо (в 50–60 % случаев) обнаруживаются неспецифические, чаще асимптомные очаги демиелинизации.
В некоторых случаях имеет место более массивное поражение головного мозга, соответствующее рассеянному склерозу.
Показатели МРТ различны при разных клинических вариантах острого демиелинизирующего энцефаломиелита и при оптикомиелите [16].
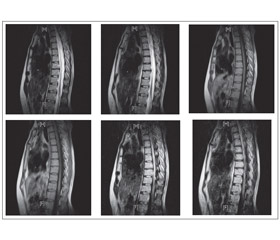
МРТ выявляет при ОНМ типичные изменения в спинном мозге. В большинстве наблюдений визуализируются очаги, распространяющиеся более чем на два позвоночных сегмента, как правило некротического характера (рис. 1).
/144/144.jpg)
Дополнительные возможности предоставляют результаты анализа ЦСЖ, ее исследование на наличие олигоклональных полос.
Дифференциальную диагностику проводят и с острым демиелинизирующим энцефаломиелитом (ОДЭМ), возникающим вскоре после перенесенной инфекции или ОРВИ и симптоматически сходным с оптикомиелитом.
В отличие от синдрома Девика у части пациентов ОДЭМ заканчивается полным выздоровлением, в некоторых случаях сохраняются остаточные явления [10].
Современные диагностические критерии оптикомиелита Девика сформулированы D. Wingerchuk [5] и включают три «абсолютных» критерия: 1 — наличие оптического неврита, 2 — наличие острого миелита, 3 — отсутствие клинических проявлений заболевания вне зрительных нервов и спинного мозга.
Кроме того, для подтверждения диагноза необходимо наличие одного из «больших» подтверждающих критериев: 1— отсутствие МРТ–изменений в головном мозге; 2 — очаговые изменения в спинном мозге протяженностью на уровне трех позвоночных сегментов и более; 3 — наличие плеоцитоза в ЦСЖ более 50 клеток в 1 мм3 или более 3 нейтрофилов в 1 мм3.
К «малым» подтверждающим критериям относятся 2 из следующих: 1 — билатеральный оптический неврит; 2 — выраженный оптический неврит со стойкой утратой зрения по крайней мере на один глаз; 3 — тяжелая стойкая остаточная слабость одной конечности и более.
Новыми критериями ОНМ являются аутоантитела — маркеры NMO–IgG. Поэтому комбинация клиники, данных МРТ и оценка NMO–IgG–статуса предоставляет максимальную диагностическую возможность по сравнению с отдельными компонентами диагноза.
В настоящее время при ОНМ допускается возможность вовлечения структур ЦНС вне зрительных нервов и спинного мозга. Есть описания клинических случаев, когда у пациентов с объективизированным диагнозом ОНМ имеется сочетание клиники оптикомиелита с асимптомными или симптомными очагами в головном мозге, например в проекции структур гипоталамо–питуитарной оси [9].
Это объясняется тем, что NMO–IgG, являясь спе–цифическими маркерами ОНМ, селективно связываются с аквапорином–4 (AQP4), концентрация которого значительно повышена в разных участках мозга. Это объясняет, почему изменения мозга вне зрительных нервов и спинного мозга локализуются в области гипоталамуса, перивентрикулярных областях — именно там, где наиболее высока экспрессия AQP4. Кроме ОНМ, высокая экспрессия этого белка определена у пациентов с эпилепсией и некоторыми видами опухолей. Предполагается, что ОНМ может представлять собой неизвестный вариант аутоиммунной каналопатии. Высказывается предположение, что следствием связывания аутоантител с AQP4 может быть прямое повреждение функции ионных каналов с развитием набухания, или антитела могут активировать комплемент, который впоследствии инициирует агрессивную воспалительную реакцию, характерную для ОНМ. Иммунологические характеристики вариантов демиелинизирующих заболеваний также различаются, что позволяет интерпретировать часть случаев как атипичный РС.
В сыворотке крови пациентов с ОНМ почти в половине случаев обнаруживаются антитела (антинуклеарные, к экстрагируемым ядерным антигенам, к двуспиральной ДНК, антитиреоидные) и их сочетания, что свидетельствует о предрасположенности этих больных с эндокринопатиями к развитию аутоиммунных заболеваний [10].
При атаке миелита с ОМ в анализе цереброспинальной жидкости у одной трети пациентов наблюдается плеоцитоз (> 50 лейкоцитов в 1 мм3) с наличием нейтрофилов и повышение уровня белка в различных пределах. Подобные изменения редко наблюдаются при РС [3]. Стратифицированный подход к лечению ОНМ в будущем может позволить систематизировать эти редкие формы демиелинизирующих заболеваний.
В настоящее время общепринятого стандарта в лечении синдрома Девика нет. Используется симптоматическое и общеукрепляющее лечение для поддержки имеющихся неврологических функций.
Возможно назначение кортикостероидов. Одним из вариантов лечения является применение препаратов, частично блокирующих В–клетки, таких как ритуксимаб. Для патогенетического лечения в мире зарегистрированы шесть препаратов, изменяющих течение заболевание. Три из них относятся к группе интерферонов; интерфероны бета–1а, бетфер–1а и интерфероны бета–1b и бетфер–1b [7].
Для лечения атаки миелита и оптического неврита применяют высокие дозы кортикостероидов. Наряду с этим эффективно использование и плазмафереза.
Эффективность превентивной иммуномодулирующей терапии у пациентов с ОНМ формально не изучена. Имеются противоречивые сведения как об эффективности интерферона бета–1, так и иммуносупрессивной терапии. Терапией выбора считают комбинацию преднизолона и аутоприна.
Лечение больных с ОНМ всегда представляет сложную задачу. Основой его является возможно более раннее начало иммуносупрессивной терапии. Клинические данные предполагают, что плазмаферез и иммуносупрессивная терапия более показаны для лечения и предупреждения атак, а стандартная иммуномодулирующая терапия не имеет существенного влияния на течение ОНМ.
Несмотря на проводимую терапию, синдром Девика во многих случаях приводит к летальному исходу [8].
Приводим собственное клиническое наблюдение больной оптикомиелитом Девика.
Больная К.Н.В., 1983 г.р., не работает.
Поступила в неврологическое отделение с жалобами на отсутствие движений в нижних конечностях, чувство онемения в нижней половине туловища и в ногах, нарушение функции тазовых органов по типу задержки.
В августе 2011 г. (спустя 6 месяцев после II родов) появились боли в грудном отделе позвоночника. Симптоматика в течение 2 недель нарастала: усилились боли в позвоночнике, появились онемение и слабость в правой ноге. 05.09.11 в сопровождении родственников обратилась за помощью в Киевскую областную больницу (в отделение сосудистой патологии и заболеваний ЦНС), где был выставлен диагноз: острый миелит грудного отдела спинного мозга на уровне Тh4–Тh8, синдром Броун–Секара (находилась на стационарном лечении). В результате проведенной терапии отмечалась положительная динамика: увеличилась сила в правой ноге до 4 баллов, восстановилась чувствительность, исчезли боли в позвоночнике. В феврале 2013 г. (во время III беременности) у пациентки наблюдалась симптоматика по типу невралгии n.trigeminus с последующим регрессом лицевой боли. Настоящее ухудшение состояния с 05.08.13 (спустя 1,5 месяца после III родов) проявилось выраженной болью в области грудного отдела позвоночника. В течение недели симптоматика прогрессировала: присоединились онемение нижней половины туловища и нижних конечностей. 10.08.13 появились выраженная слабость в нижних конечностях, нарушение функции тазовых органов по типу задержки, и 11.08.13 пациентка в сопровождении родственников была ургентно доставлена в приемное отделение ОКБ.
Неврологический статус (на момент осмотра в неврологическом отделении): сознание ясное, астенизирована, эмоционально лабильна, фиксирована на своих ощущениях. Глазные щели D = S, зрачки D = S, фотореакции их живые, офтальмодинамика не нарушена. Лицо симметрично. Активные движения в руках в полном объеме; в ногах движения отсутствуют (нижняя параплегия), мышечный тонус в ногах повышен по пирамидному типу, (+) симптом Бабинского с 2 сторон. Сухожильные рефлексы с верхних конечностей оживленные, D = S; высокие, с нижних D = S. Гипестезия по проводниковому типу с уровня Тh4. Нарушение функции тазовых органов по типу задержки.
Обследования
РВ от 11.04.13: отриц.
ОАК от 13.08.13: Э 4,01 Т/л, Л 6,6 Г/л, Нb 105 г/л; СОЭ 11 мм/ч; п 1, с 78, л 10, м 11; анизоцитоз слабо выражен.
ОАК от 27.08.13: Э 4,10 Т/л, Л 7,6 Г/л, Нb 108 г/л; СОЭ 14 мм/ч; э 2, п 7, б 1, м 4, л 22, с 64; анизоцитоз.
Анализ крови на LE–клетки от 14.08.13: не обнаружены.
Коагулограмма от 27.08.13: ПТВ 18”; ПТИ 80,5 %; МНО 1,31; АЧТВ 28”.
Пробы на активность ревматизма от 14.08.13: СРБ (=); РФ (–); антистрептолизин–О (–); серомукоиды — 0,29.
РГ ОГК от 11.08.13: патологических изменений со стороны органов грудной клетки не выявлено.
Антитела к аквапорину–4 от 15.08.13: аnti–aquaporin–4 IgA, G, M (IgA — отриц., IgG — +++ 1 : 320, IgM — отриц.); серологический диагноз: Neuromyelitis optica (NMO), оптико–спинальный энцефаломиелит, синдром Девика Anti–Hu — отриц. Anti–Ri — отриц. Anti–Yo — отриц. Anti–Tr — отриц. Anti–MAG — отриц. Anti–Myelin — отриц. Anti–Ma/Ta — отриц. Anti–GAD — отриц. Anti–Amphiphysin — отриц. Anti–GlutamatRezeptoren — отриц. Anti–Gaba–b–Rezetoren — отриц. Anti–LGIl — отриц. Anti–CASPR2 — отриц. Anti–Glycinrezeptoren — отриц.
Анализ крови на TORCH–инфекции от 15.08.13: антитела IgG к CMV — 2,53; антитела IgG к HSV — 3,65, антитела IgG к Rubella — 3,36, антитела IgG к Toxoplasma gondii — 2,57; антитела IgM к HSV — 0,52, антитела IgM к Rubella — 0,07, антитела IgM к Toxoplasma gondii — 0,13.
MPT головного мозга от 12.08.13: объемных и очаговых изменений со стороны структур головного мозга не выявлено.
МРТ грудного отдела позвоночника от 12.08.13: на серии сагиттальных, коронарных и аксиальных срезов грудного отдела позвоночника в Т1– и Т2–взвешенных изображениях при сравнении с МТР–исследованием от 02.09.11 в структуре спинного мозга на уровне Тh4–Тh9 (до верхнего края тела Тh9 позвонка) сохраняется центрально расположенная зона патологического изменения интенсивности сигнала, распространяющаяся практически на всю толщину спинного мозга, гиперинтенсивного характера в Т2–ВИ, STIR, изогиперинтенсивного характера в последовательности FLAIR. На уровне Тh11–Тh12 и в проекции эпиконуса визуализируется еще один очаг гиперинтенсивного характера в Т2–ВИ, STIR, гипоинтенсивного характера в Т1–ВИ, который определялся и на предыдущих снимках. Заключение: изменения в структуре спинного мозга могут быть обусловлены диффузной формой поперечного миелита. При сравнении с МРТ–исследованием от 02.09.11 г. отмечается некоторое прогрессирование основного заболевания. Рекомендовано внутривенное контрастное усиление, пациентка от введения контрастного вещества отказалась. Признаки дегенеративно–дистрофического процесса по типу остео–хондроза. Дископатия на уровне Th7–Th8.
Гематолог от 16.08.13: диагноз: анемия, обусловленная неврологической патологией и после беременности. Рекомендовано: гино–тардиферон по 1 табл. 1 р. в день 1 месяц; препараты витаминов группы В; контроль показателей периферической крови в динамике.
Гинеколог от 12.08.13: гинекологической патологии не выявлено.
Окулист от 12.08.13: диагноз: атрофия зрительных нервов.
Зрительные ВП 12.08.13 — снижена амплитуда ответа с деформацией комплекса ЗВП симметрично. Выявлены признаки аксонального поражения обоих зрительных нервов.
Уролог от 14.08.13: нейрогенный мочевой пузырь по гипотоническому типу, на фоне миелита. Рекомендовано: омникс по 1 к. на ночь 1 месяц; но–шпа по 1 табл. 3 р. в день 10 дней; свечи диклоберл 50 мг по 1 св. 1 р. в день 10 дней; палин по 2 табл. 2 р. в день 14 дней; промывать уретральный катетер стерильным физраствором 10,0 мл + декасан 10,0 мл 2 р. в день. Уролог от 06.09.13: нейрогенный мочевой пузырь. Острая задержка мочи.
Рекомендовано: установить постоянный уретральный катетер; уход за катетером — декасан 4 мл + 16 мл физ. р–ра — промывать 1 р. в день; канефрон по 2 табл. 3 р. в день 1 месяц. Проведенное лечение: солу–медрол (1000 мг в/в кап. в течение 7 дней); L–лизина эсцинат, глиатилин, нейробион, эссенциале, сульфокамфокаин, прозерин, АТФ, анальгин, димедрол, цефатоксим, омникс, алора, гербастресс, достинекс, омез, келтикан, тонгинал, АТФ–лонг. ЛФК, массаж нижних конечностей. ЭФ с прозерином на область мочевого пузыря; электростимуляция нижних конечностей, плазмаферез (3 сеанса).
За время пребывания в стационаре непереносимости к медпрепаратам не отмечалось.
В результате проведенного лечения отмечается некоторая положительная динамика в состоянии пациентки: появились минимальные движения в дистальных отделах нижних конечностей (движения в левой стопе и в пальцах правой стопы), начала самостоятельно садиться, поворачивается в постели, появились позывы к мочеиспусканию и дефекации, восстановилась чувствительность в нижних конечностях до уровня верхней трети бедер.
Имеющаяся у нашей пациентки неврологическая симптоматика вполне определенно характеризует поражение зрительных нервов и грудных сегментов Th11–Th12 спинного мозга. Наличие антител к аквапорину–4 свидетельствует в пользу активного аутоиммунного процесса. Клиническое течение заболевания оценено как ремиттирующее, поскольку очередная атака ОНМ возникала в послеродовом периоде и сопровождалась поражением тех же органов, которое подтверждалось при нейровизуализации.
В стационаре была проведена дифференциация с ремиттирующим течением спинальной формы РС, острым некротическим поперечным миелитом Алажуанина. В пользу РС свидетельствовал дебют и экзацербация процесса после родов, наличие 2 очагов поражения (зрительные нервы и спинной мозг), а также преходящий характер прозопалгии, которая регрессировала самостоятельно.
Против РС могут свидетельствовать такие признаки, как поперечный миелит, который, по данным МРТ спинного мозга, распространялся классически — продольно на 2–3 сегмента, отсутствие изменений на МРТ головного мозга, а также наличие антител к аквапорину–4 IgG.
В пользу острого некротического поперечного миелита Фуа — Алажуанина свидетельствовало также острое развитие заболевания, характеризующееся поперечным поражением грудного отдела спинного мозга с развитием соответствующих данному уровню поражения спинальных сенсомоторных и сфинктерных расстройств.
Кроме того, при некротическом поперечном миелите наблюдаются разнообразные варианты сенсорных расстройств типа синдрома Броун–Секара, заднестолбового, спиноталамического характера, ранние трофические расстройства в виде пролежней, воспалительный характер цереброспинальной жидкости, возможно и крови.
Кроме того, поражение зрительных нервов нетипично для данного заболевания.
С учетом того, что у нашей пациентки наряду с атрофией зрительных нервов отсутствовали воспалительные изменения в ЦСЖ и периферической крови, заднестолбовой и сегментарный типы сенсорных расстройств, пролежни, диагноз поперечного миелита был снят.
Несомненно то, что исходным диагнозом в настоящее время является оптиконевромиелит (болезнь Девика). Однако, учитывая общие аутоиммунные патогенетические механизмы данного заболевания и РС, исключить возможность трансформации в РС не представляется возможным.
В связи с этим обоснованным является динамическое наблюдение за пациенткой и соответствующий иммунологический контроль.
Клинический диагноз может быть сформулирован следующим образом: оптиконевромиелит (болезнь Девика), ремиттирующее течение, экзацербация, нижняя спастическая параплегия, нарушение функции тазовых органов по типу задержки, атрофия зрительных нервов обоих глаз.
1. Бушуева H.H. Синдром Девика (оптикомиелит) при рассеянном склерозе / H.H. Бушуева, Е.С. Стоянова // Офтальмологический журнал. — 2012. — № 3. — С. 83–86.
2. Богданов Э.И., Михайлов И.М., Селезнева А.В. Оптикомиелит // Неврологический журнал. — 2004. — Т. 9, № 2. — C. 17–21.
3. Евтушенко С.К., Москаленко М.А. Рассеянный склероз у детей. — К.: Здоров’я, 2008. — 356 с.
4. Журавлев М.Н., Сиверцева С.А., Молокова М.Ю. и др. Оптикомиелит, или болезнь Девика: патогенез, клиника, диагностика и опыт лечения с использованием бетаферона и митроксана // Журнал неврологии и психиатрии им. С.С. Корсакова. — 2007. — Вып. 4. – С. 106–112.
5. Яхно Н.И., Мозолевский Ю.В., Голубева В.В. и др. Оптикомиелит Девика // Неврологический журнал. — 2008. — Т. 13, № 2. — ISSN 1560–9545. — C. 27–32.
6. Шахов Б.Е., Белова А.Н., Шаленков И.В. Оптикомиелит Девика: вопросы диагностики и описание случая из практики // Медицинский альманах. — 2012. — № 1 (20). — С. 165–170.
7. Barrer J., Metz Z. Devic’s neuromylitis optica treated with intravenous gammaglobulin (IVIG) // Can. J. Neurol. Sci. — 2004. — Vol. 31. — P. 265–267.
8. Cabrera–Yomez J.A., Quevedo–Sotolongo Z. Brain magnetic resonance imaging findings in relasping neuromyelitis optica // Mult. Scler. — 2007. — Vol. 13. — P. 186–192.
9. Chalumеau–Zemoine Z., Chretien F., Si Zarbi A.Y. Devic disease with brainstem lesion // Arch. Neurol. — 2006. — Vol. 63. — P. 591–593.
10. De Seze J., Storovic T., Ferriby D. Devic’s neuromyelitis optica: clinical, laboratory MRI and outcome protile // J. Neurol. Sci. — 2002. — № 197. — Р. 57–61.
11. Giovannoni G. Neuromyelitis optics and anti–aquaporin–4 antibodies: widening the clinical phenotype // J. Neurol. Neurosung Psichiatry. — 2006. — Vol. 77. — P. 1073–1075.
12. Maticlo M., Weinshshenker B. Neuromyelitis optica // C.F. Zucchinetti. Multiple sclerosis. — 2010. — P. 258–275.
13. Minagar A., Alexander J.S., Fowler M.R. Device disease: clinical course, pathophysiology and management // Phathophysio–logy. — 2002. — Vol. 3. — Р. 33–40.
14. Miyazava I., Fujihara K., Itoyama G. Eugene Devic (1858–1930) // J. Neurol. — 2002. — Vol. 3. — P. 351–352.
15. Pittock S.Y., Zennon V.A., Krecke K. Brain abnormalites in neuromyelitis optica // Arch. Neurol. — 2006. — Vol. 63. — Р. 390–396.
16. Rossa M.A., Agosta F., Mzzapesa D.M. Magnetization transber and difusion tensor MRS show gray matter damage in neuromyelitis optica // Neurology. — 2004. — Vol. 32. — P. 476–478.