
Газета «Новости медицины и фармации» Аллергология (345) 2010 (тематический номер)
Вернуться к номеру
Аллергические «маски» соматических болезней у детей
Авторы: Аллергические «маски» соматических болезней у детей
Версия для печати
Первичные иммунодефициты имеют много клинических проявлений, напоминающих аллергические заболевания. Прежде всего это касается гуморальных иммунодефицитов и некоторых форм комбинированной иммунной недостаточности.
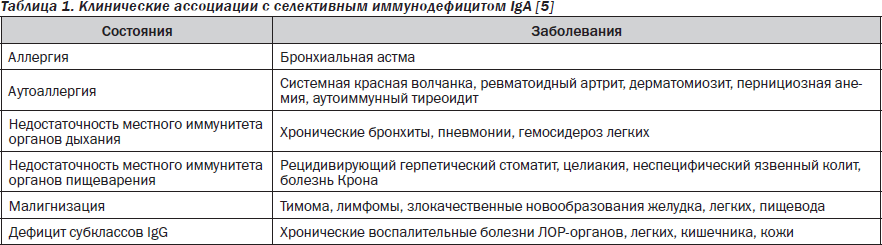
Селективный иммунодефицит ІgA, в т.ч. селективный дефицит секреторного ІgA, является наиболее частой первичной аномалией иммунной системы — его распространенность составляет 1 : 100 – 1 : 700 среди детей европейской популяции [5, 17]. Этот вариант первичного дефицита гуморального звена иммунитета характеризуется низким содержанием сывороточного IgА — 0,05 г/л у детей старше 2 лет. Есть 4 основных варианта селективного дефицита ІgA: без клинических проявлений, при атаксии-телеангиэктазии (первичном комбинированном иммунодефиците — синдроме Луи-Бар), в сочетании с гипер-ІgМ, в сочетании с хромосомными мутациями [5, 17]. Поскольку IgА, особенно его секреторный компонент, является главным фактором системы местного иммунитета, самыми частыми клиническими проявлениями являются рецидивирующая и хроническая патология дыхательных путей и ЛОР-органов, а также дисбиоз кишечника и другие поражения органов пищеварения. При секреторном дефиците IgА развиваются аллергические и аутоиммунные процессы (табл. 1).
Следует отметить, что приблизительно 40 % детей с сывороточным дефицитом IgА имеют антитела против IgG, что создает предпосылки для развития анафилактоидных реакций во время гемотрансфузий и переливания препаратов крови. Весьма часто обнаруживаются антитела к белкам коровьего молока и куриного яйца, мяса, что клинически проявляется симптомами пищевой аллергии [5, 17].
Дифференциальным признаком иммунодефицита IgА является низкий его уровень в крови и/или секретах. Содержание Т-лимфоцитов, их популяций и В-лимфоцитов нормальное.
Первичная агаммаглобулинемия (болезнь Брутона) — Х-сцепленное рецессивно наследуемое заболевание, характеризующееся снижением общего содержания гамма-глобулинов сыворотки крови до 2 г/л и меньше за счет IgG, IgА и IgМ. При этом варианте врожденного иммунодефицита снижена резистентность к стафилококку, стрептококку, пневмококку, а также к кишечной палочке, сальмонелле, протею, клебсиелле [5, 17]. Дети часто болеют рецидивирующими пневмониями, отитами, пиодермиями, которые могут приводить к развитию сепсиса. Часто наблюдаются грибковые поражения и пневмоцистные пневмонии, однако к вирусам отмечается нормальный иммунный ответ, и некоторые вирусные инфекции (краснуха, корь, вирусный гепатит) у больных протекают даже легче, чем у детей с нормальным возрастным иммунитетом. Однако к вирусу полиомиелита дети проявляют высокую чувствительность [17]. Кроме воспалительных процессов, у детей старше 3 лет отмечаются аутоиммунные болезни (дерматомиозит, полиартриты). Характерной особенностью этого иммунодефицита является отсутствие аденоидных вегетаций и гипертрофии миндалин, а также не наблюдается увеличения лимфоузлов, печени и селезенки даже во время обострения воспалительного процесса. У некоторых детей отмечаются аллергические реакции на медикаменты (антибиотики). Воспалительные процессы сочетаются с атопической патологией — атопическим дерматитом, аллергическим ринитом и бронхиальной астмой, которые отмечаются у 40 % больных. Это обусловлено тем, что синтез IgЕ сохранен. Туберкулиновые пробы и реакция БЦЖ у больных нормальные.
Синдром гипериммуноглобулинемии Е (гипер-ІgЕ-синдром, синдром золотистого стафилококка с гипер-ІgЕ, синдром Ние) — симптомокомплекс, характеризующийся увеличением в сыворотке крови ІgЕ, что сопровождается развитием атопического дерматита, подкожных абсцессов преимущественно стафилококковой природы и другими рецидивирующими инфекционными процессами. Впервые клиническую картину (образование «холодных» стафилококковых абсцессов) описал в 1966 году S.D. Davis и соавт. под названием «синдром Джоба». Наиболее частыми возбудителями инфекционных процессов являются Staphylococcus aureus и Candida albicans. Заболевание начинается обычно с первых месяцев жизни. В клинике доминирует пиодермия с генерализованным экзематозным дерматитом с поражением лица, волосистой части головы и шеи с сильным зудом [2, 15]. Нередко экзема сопровождается другими атопическими заболеваниями — бронхиальной астмой, отеком Квинке, аллергическим ринитом. В дальнейшем присоединяются рецидивирующие инфекции в виде гнойного среднего отита, ринита, стафилококковой пневмонии, подкожных «холодных» абсцессов без классических признаков воспаления, кандидоза слизистых оболочек и кожи, сепсиса. Характерна эозинофилия различной степени. Диагноз устанавливается на основании анамнеза, типичной клинической картины, лабораторных данных: высокого содержания общего ІgЕ в крови, значительных титров антистафилококковых ІgЕ-антител, угнетения хемотаксиса нейтрофилов, недостаточности фагоцитоза [2, 15].
Синдром Вискотта — Олдрича — Х-сцепленный первичный комбинированный иммунодефицит, характеризующийся тромбоцитопенией, экземой, нарушением клеточного и гуморального звеньев иммунитета, склонностью к лимфопролиферативным заболеваниям. До недавнего времени считалось, что синдром Вискотта — Олдрича встречается у мальчиков, однако в последнее время описано несколько случаев и у девочек. Для этого заболевания характерно отсутствие В-лимфоцитов, продуцирующих ІgM, содержание же ІgG, как правило, не изменено, а концентрация ІgА, ІgЕ и ІgЕ — увеличена [5, 17]. В результате комбинированного иммунодефицита и резкого снижения резистентности к инфекциям у больных наблюдаются хронические и рецидивирующие вирусные и бактериальные инфекции, трудно поддающиеся терапии с применением традиционных противоаллергических мероприятий. Чаще всего отмечаются отиты, пиодермии, колиты. Нередко присоединяются грибковые поражения и пневмоцистная пневмония. Наблюдаются нарушения гемостаза в виде удлиненного времени кровотечения, нарушения рефракции кровяного сгустка. Прогноз в целом неблагоприятен. При тяжелых формах дети погибают в течение первых 10 лет от инфекций и дистрофии. При менее тяжелых вариантах болезни продолжительность жизни может быть большей, однако с возрастом существенно увеличивается риск развития злокачественных новообразований, особенно неходжкинских лимфом [5, 17].
Диагностика синдрома Вискотта — Олдрича достаточно сложна из-за сходства симптомов с другими первичными иммунодефицитами и аллергическими болезнями. Дифференциально значимым признаком является низкий уровень сывороточного ІgM на фоне гипериммуноглобулинемии А и Е. У больных не синтезируются антитела к полисахаридам и белковые антигены, наблюдается кожная анергия. Количество Т-лимфоцитов прогрессивно уменьшается, в то время как содержание В-лимфоцитов увеличивается. При анализе гемограммы следует обращать внимание на уровень и размер тромбоцитов. Наиболее точный диагноз устанавливается при хромосомном анализе с применением FISH-технологии [5].
Ангионевротический отек (АО) — заболевание, характеризующееся появлением отека кожи, подкожной клетчатки или слизистых оболочек, который остро развивается и относительно быстро проходит. Есть формы ангионевротического отека, обусловленные патологией системы комплемента как наследственного, так и приобретенного характера [6]:
1. Наследственная форма — первичный иммунодефицит, обусловленный расстройствами в системе комплемента:
— тип І (аутосомно-доминантное наследование): уровень С1-ингибитора составляет 0–30 % от нормального;
— тип ІІ (аутосомно-доминантное наследование): функциональная недостаточность С1-ингибитора, уровень С1-ингибитора нормальный;
— тип ІІІ (сцепленное с Х-хромосомой аутосомно-доминантное наследование): уровень С1-ингибитора и его функция нормальны. Наблюдается только у лиц женского пола.
2. Приобретенная форма:
— тип 1: дефицит С1-ингибитора у больных с лимфопролиферативными заболеваниями;
— тип 2 характеризуется наличием аутоантител к С1-ингибитору при гетерогенной патологии (диффузные заболевания соединительной ткани, онкологическая патология, болезни печени), а также у лиц без признаков каких-либо заболеваний.
3. Другие формы ангионевротического отека.
У больных ангионевротическим отеком за счет дефектов С1-ингибитора отмечаются, в отличие от отека Квинке, такие характерные черты [4, 6, 11]:
— доступный осмотру отек бледный, очень плотный, зуд отсутствует; при надавливании на него не остается ямки;
— развитие отека медленное, в течение нескольких часов;
— крапивница отсутствует;
— во время оперативного вмешательства по поводу «острого живота» у больного обнаруживают отек участка кишки и асцитический выпот;
— при отеке мочевыводящих путей отмечается задержка мочи;
— сильная головная боль наблюдается при отеке мозговых оболочек;
— возможен отек гортани.
Необходимо исследовать в сыворотке крови уровни С4-компонента комплемента и С1-ингибитора. Содержание общего IgE в крови нормальное. Применение кортикостероидов и Н1-антигистаминов не эффективно [4, 6].
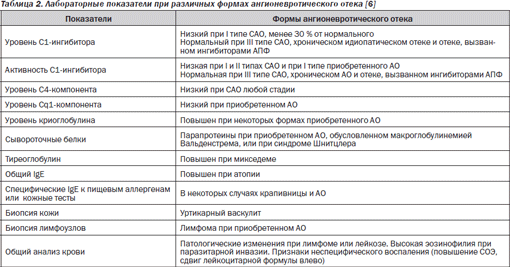
В табл. 2 приведены дифференциально-диагностические критерии различных вариантов ангионевротического отека.
Значительную сложность для аллерголога представляют вопросы дифференциальной диагностики различных проявлений кожного синдрома. Это в первую очередь касается системных васкулитов, ревматической лихорадки и диффузных болезней соединительной ткани.
Системные васкулиты — группа заболеваний, характеризующихся первичным поражением стенки сосудов различного калибра по типу очагового воспаления и некроза и вторичным повреждением органов и тканей зоны сосудистого поражения. Известно, что ведущим патогенетическим механизмом заболеваний этой группы является образование большого количества иммунных комплексов. У детей встречаются такие системные васкулиты: геморрагический васкулит, слизисто-кожно-лимфонодулярный синдром Кавасаки, узелковый полиартериит, неспецифический аортоартериит, гранулематоз Вегенера, эозинофильный гранулематозный ангиит (синдром Чарга — Стросса (СЧС)) и синдром Бехчета. В дебюте большинства системных васкулитов наблюдаются общие черты неспецифического воспалительного синдрома: субфебрилитет или фебрилитет, артралгии, исхудание, симптомы периферических или висцеральных сосудистых нарушений, признаки воспаления по лабораторным данным. Наряду с этим каждое заболевание имеет характерную клиническую симптоматику (табл. 3).

Геморрагический васкулит (болезнь Шенляйн — Геноха) — распространенная форма васкулитов с подавляющим поражением микроциркуляторного русла. Клиническая картина разноплановая, может быть представлена одним кожным синдромом или его сочетанием с другими характерными синдромами: абдоминальным, суставным, почечным. Кожный синдром может проявляться симметричной полиморфной сыпью — мелкопятнистой, папулезной, уртикарной, но без зуда — с излюбленной локализацией вокруг суставов, на разгибательных поверхностях конечностей. Однако патогномоничным кожным симптомом является геморрагическая пурпура, локализующаяся на папулах и уртикариях, часто с формированием некротических корок [1, 8, 16]. Вариантом кожного синдрома является простая кожная форма с «холодовой» крапивницей и «холодовым» отеком Квинке, которые локализуются на различных участках конечностей, туловища и даже головы и характеризуются летучестью, однако не имеют признаков типичного воспаления. У части детей развитие этих признаков обусловлено не гистаминовым типом реакций, а расстройствами в системе комплемента и действием других биологически активных веществ, поэтому Н1-антигистамины в этом случае не эффективны. Стоит напомнить, что до манифестации геморрагической пурпуры могут быть повторные эпизоды пятнисто-папулезной или уртикарной сыпи, что часто расценивается как проявления аллергического процесса.
Слизисто-кожно-лимфонодулярный синдром (болезнь Кавасаки) — острая безрецидивная форма системных васкулитов с преобладающим поражением мелких и средних артерий и морфологией, аналогичной узелковому полиартерииту. Наблюдается преимущественно у детей до 8 лет, чаще у мальчиков первого года жизни. Клинически проявляется высокой температурой в течение 12–36 суток, гиперемией конъюнктив, инъекцией склер, диффузной гиперемией слизистой оболочки ротоглотки, «малиновым» языком, интенсивной эритемой кожи или полиморфной сыпью с эволюцией в шелушение кожи кончиков пальцев рук в конце 2-й — начале 3-й недели болезни [8]. Такая клиническая симптоматика напоминает некоторые детские инфекции — корь, краснуху и скарлатину, однако, учитывая формирование отеков на нижних и верхних конечностях, затяжной и распространенный характер высыпаний (на лице, туловище, конечностях), возникает потребность дифференциации с аллергическим заболеванием. Нередко имеют место цианотичные отеки кистей и стоп, артралгии, диарея, увеличение печени. Помогает в постановке правильного диагноза наличие увеличения лимфоузлов шейной группы более 1,5 см в диаметре, изменений со стороны сердца — кардиомегалии, нарушения ритма сердца, систолического шума. Важное диагностическое значение имеет характерное для синдрома Кавасаки развитие коронарита, обнаруживаемого с помощью ЭКГ, коронарографии и ЭхоКГ. В большинстве случаев прогноз болезни Кавасаки благоприятный, однако поражение коронарных сосудов является фактором риска неблагоприятного ближайшего и отдаленного прогноза, обусловленного разрывом коронарной аневризмы, развитием ранней ишемической болезни сердца, инфаркта миокарда [8].
Узелковый полиартериит — системный васкулит с поражением периферических и висцеральных артерий преимущественно мелкого и среднего калибра — встречается у детей разного возраста, чаще у девочек. Заболевание начинается в большинстве случаев остро: отмечаются высокий фебрилите, профузная потливость, интенсивные миалгии, артралгии, абдоминалгии, значительное исхудание [1, 8]. Через несколько недель появляются характерные клинические признаки (табл. 4). Иногда за какое-то время до клинической манифестации узелкового артериита отмечается эпизод геморрагического васкулита.

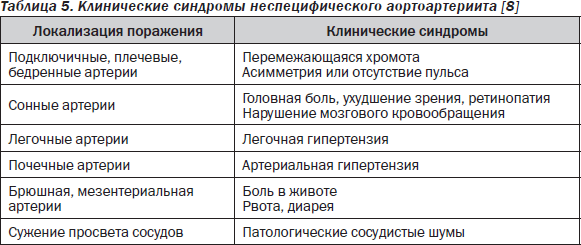
Неспецифический аортоартериит (НАА, болезнь отсутствия пульса, синдром Такаясу) — деструктивно-пролиферативный сегментарный аортит и субаортальный панартериит — характеризуется образованием аневризм и/или стенозов аорты или ее ветвей, даже сегментарной артериальной окклюзии, что клинически проявляется ишемическими расстройствами и синдромом асимметрии или отсутствия пульса [8, 10]. Могут поражаться дуга аорты или ее ветви (сонная, брахицефальная, подключичная), грудная и брюшная аорта и артерии, отходящие от них (мезентериальные, почечные и др.). Возможно сочетание этих вариантов с поражением легочной артерии. Болезнь встречается преимущественно у лиц женского пола, начинается в возрасте 8–20 лет, у младших детей наблюдается редко. В дебюте НАА отмечается лихорадка, миалгии, реже — геморрагические или узловатые высыпания, стойкое и значительное увеличение СОЭ. Синдром асимметрии и болезнь отсутствия пульса регистрируются спустя 1–5 лет от начала болезни. Основные проявления НАА представлены в табл. 5.
Синдром Чарга — Стросса — системный некротизирующий васкулит с поражением сосудов мелкого и среднего калибра, с гранулематозным воспалением легочной системы при наличии астмы и эозинофилии. В 1951 году J. Churg и L. Strauss описали 13 случаев диссеминированного некротизирующего васкулита у пациентов с тяжелой астмой, лихорадкой и эозинофилией, а в 1994 году на Согласительной конференции по номенклатуре системных васкулитов была определена его принадлежность к этой группе заболеваний. Классический СЧС начинается с поражения верхних дыхательных путей — аллергического ринита, назального полипоза или синусита [8, 12]. Одновременно или позже развивается бронхиальная астма, которая у большинства больных является основным клиническим синдромом на протяжении нескольких лет. В 38–77 % случаев обнаруживают транзиторный легочный инфильтрат. У трети больных присутствует плеврит с эозинофилией в плевральной жидкости. В дальнейшем развивается прогрессирующая потеря массы тела, лихорадка, астения, артралгии, иногда артриты, миалгии, поражения кожи — геморрагическая пурпура, эритема, крапивница, кожные некрозы, сетчатое ливедо, подкожные узелки [14, 19–21]. Следующий этап заболевания — генерализация, развитие системного васкулита, при этом степень тяжести бронхообструктивного синдрома по большей части уменьшается. Среди прогностически значимых висцеральных проявлений чаще (36–62 %) всего встречается поражение органов пищеварения — гастроэнтерит с эозинофильной инфильтрацией или васкулитом брыжеечных сосудов. В клинике доминируют абдоминалгии, реже — тошнота, рвота диарея, кровотечение, мелена. Осложнениями васкулита являются образование язв желудка или кишечника, кровотечение, перфорация, перитонит, встречаются некротические и ишемические повреждения органов пищеварения с развитием инфарктов, васкулиты поджелудочной желе- зы, желчного пузыря, печенки [20–22].
Периферическая нервная система поражается у 64–75 % больных. Чаще всего наблюдается множественный мононеврит, реже — полинейропатия по типу «перчаток и носков», дебютирующая множественным мононевритом. В классических случаях больные жалуются на боль, слабость и снижение чувствительности в зоне иннервации пораженного нерва. Чаще поражаются 2 и больше нервных ствола.
Симптомы поражения центральной нервной системы наблюдаются у 3–7 % больных и проявляются энцефалопатией, инсультами, субарахноидальным кровоизлиянием, эпилептиформными судорогами, гиперкинезами или психическими расстройствами [8]. Развивается геморрагический или ишемический инсульт, как правило обусловленный цереброваскулитом, а в некоторых случаях — артериальной гипертензией.
Поражение сердечно-сосудистой системы встречается в 15–64 % случаев и клинически характеризуется перикардитом, миокардитом, эндокардитом, коронаритом, инфарктом миокарда, обусловленным коронаритом [12, 14, 19].
Поражение почек отмечается у 15–88 % больных. При этом наблюдается протеинурия, гематурия, признаки почечной недостаточности, а также повышение артериального давления.
Диагностика СЧС базируется на выявлении эозинофилии (на любой стадии заболевания), однако встречаются случаи без периферической эозинофилии с выраженной тканевой эозинофильной инфильтрацией. Характерным признаком является наличие антител к миелопероксидазе нейтрофилов (МРО-АNCA) у 48–66 % больных. Подтверждают диагноз и морфологические исследования.
Диффузные болезни соединительной ткани также имеют некоторые схожие иммунопатологические механизмы и клинические черты аллергической патологии.
Системная красная волчанка (СКВ) — системное аутоиммунное заболевание неизвестной этиологии, в основе которого лежит генетически обусловленное нарушение иммунорегуляторных механизмов, определяющих продукцию широкого спектра органонеспецифических аутоантител к различным компонентам ядра клеток и формирование иммунных комплексов, вызывающих развитие иммунного воспаления в тканях различных органов. СКВ характеризуется генерализованным поражением микроциркуляторного русла и системной дезорганизацией соединительной ткани с кожными, суставными и висцеральными поражениями [3, 13].
Кожный синдром наблюдается у 97 % больных и проявляется весьма полиморфными элементами, но без зуда [13]:
— эритематозные высыпания на лице в области скуловых дуг и переносицы (волчаночная «бабочка»), возможна сыпь на открытых участках тела — в области декольте, реже — над крупными суставами, иногда с появлением типичных элементов сыпи отмечаются эпизоды рецидивирующей крапивницы или крапивницы хронического течения, не поддающейся лечению Н1-антигистаминами;
— дискоидные эритематозные очаги с гиперемией, инфильтрацией, фолликулярным гиперкератозом и последующей рубцовой атрофией;
— фотосенсибилизация — повышенная чувствительность кожи к инсоляции, влиянию УФО, проявляется усиленным высыпанием или его появлением после пребывания на солнце;
— капиллярит (отечная эритема с телеангиэктазиями и атрофией) ладоней и подошвенной поверхности стоп, сетчатое ливедо (сетчатые цианотично-фиолетовые пятна на коже нижних и верхних конечностей и туловища, обусловленные застоем крови в капиллярах или микротромбозом венул), пурпура, подногтевое кровоизлияние, феномен Рейно;
— алопеция очаговая или диффузная.
Для поражения слизистых оболочек характерны:
— хейлит (поражение красной каймы губ);
— энантема (эритематозно-отечные пятна с четкими границами, а иногда с эрозивным центром, находящимся в области твердого неба);
— афтозный стоматит (эрозивные или язвенные очаги с интенсивной эритемой).
Следует отметить, что клинические и иммунологические проявления СКВ могут имитировать клиническую картину медикаментозной волчанки (волчаночноподобного синдрома) в результате действия некоторых лекарств (гидралазин, изониазид, прокаинамид, пенициллины, сульфаниламид, метилдопа, противосудорожные препараты, хлорпромазин и т.п.). Для него характерными являются лихорадка, артрит, полисерозит, сыпь. Указанные симптомы возникают на фоне приема препарата и исчезают после его отмены [13].
Ювенильный ревматоидный артрит (ЮРА), системный вариант с олигоартритом или отсроченным суставным синдромом (ранее называемый аллергосептическим вариантом ЮРА) — один из самых распространенных вариантов дебюта висцеральной формы этого заболевания, что всегда проявляется кожным синдромом. Сыпь характеризуется пятнистыми и/или папулезными элементами линейного типа, без зуда. Она не стойкая, то появляется, то исчезает на протяжении короткого времени, усиливается на высоте лихорадки, локализуется в области суставов, на лице, на боковых поверхностях туловища, ягодицах и конечностях [13]. В некоторых случаях может быть уртикарная или геморрагическая сыпь.
Ювенильный дерматомиозит (ЮДМ) с васкулитом — тяжелое прогрессирующее системное заболевание мышц, кожных покровов и сосудов микроциркуляторного русла с менее четким поражением внутренних органов, нередко осложняющееся кальцинозом и гнойной инфекцией.
При этом заболевании кожный синдром в виде дерматита на открытых частях тела является постоянным симптомом болезни. В отличие от СКВ эритема имеет цианотичный оттенок (цвет «гелиотропа», отсюда другое название ЮДМ — «лиловая болезнь»), локализуется на лице в виде периорбитальных «очков», на ушах, над суставами. Эритема может сопровождаться инфильтрацией, гиперкератозом или истончением кожи. Нередко она сочетается с отеками губ, ушных раковин, периорбитальной клетчатки. Сыпь над межфаланговыми и пястнофаланговыми суставами кистей (синдром Готтрона) оставляет после себя (чаще через 1–2 года) депигментированные атрофические рубцы, типичные для ЮДМ и являющиеся визитной карточкой болезни [2, 9]. Сквамозный характер сыпи в некоторых случаях наводит на мысль об аллергических заболеваниях и псориазе. Сыпь может быть полиморфной: одновременно на коже есть различные элементы и очаги гипо- и гиперпигментации (пойкилодермия). Возможна очаговая или тотальная алопеция и дистрофия ногтей.
Сосудистый компонент представлен капилляритами ладоней, сетчатым ливедо на груди, спине, в подмышечных участках и на конечностях, а в более позднем периоде — телеангиэктазиями чаще на верхних веках или в зоне ногтевого ложа. Генерализованное поражение сосудов, характерное для детей дошкольного возраста, часто сопровождается болью в пораженных зонах, некрозами, язвенными и гнойными процессами [9].
Поражение слизистых оболочек с развитием гиперемии и точечных кровоизлияний встречается в виде хейлита, гингивита, стоматита и даже эрозивно-язвенного эзофагита, реже страдает желудок и кишечник. Возможны катаральный и субатрофический ринит, конъюнктивит, вульвовагинит.
В дифференциальной диагностике диффузных заболеваний соединительной ткани и аллергической патологии большое значение имеет, кроме особенностей кожного синдрома, появление прогрессирующей слабости, потери аппетита, прогрессирующей дистрофии, интермиттирующей лихорадки, полилимфаденопатии, умеренного гепатолиенального синдрома, многообразных висцеральных поражений (со стороны сердца, легких, почек, полисерозита), типичных проявлений суставного и миопатического синдромов, поражения глаз в сочетании с характерными лабораторными признаками (анемия, разнонаправленые изменения) уровня лейкоцитов, тромбоцитов, лейкоцитарной формулы, биохимических параметров, наличие повышенного уровня ЦИК и иммунологических критериев: для СКВ — снижение титра общего комплемента и его компонентов (С3, С4), наличие антинуклеарного фактора (АНФ), антител к двухспиральной ДНК, антител к РНК-молекулам — к Sm-Aг, антифосфолипидных антител — к кардиолипину, фосфатидилсерину, β2-гликопротеина 1, наличие волчаночного антикоагулянта, ложноположительная реакция Вассермана, наличие антигенов HLA-A1, HLA-B8, HLA-DR2, HLA-DR3, HLA-DQw1, HLA-DQw2; для ЮРА — наличие АНФ, иногда позитивный ревматоидный фактор, повышение уровня IgG и IgM, наличие антигенов НLA-DR4 HLA-A2, HLA-B27; для ЮДМ — увеличение активности трансферраз, лактатдегидрогеназы, альдолазы, креатинфосфокиназы, повышение миоглобина крови, креатинурия, наличие миозитспецифических антител — анти-Jo-1, анти-Mi-2, анти-PM-1, SPR, а также антигенов HLA-B8, HLA-B14, HLA-DR3) [2, 9, 13].
Синдромы гиперэозинофилии. Известно, что поздние реакции гиперчувствительности немедленного типа обусловлены высвобождением цитокинов из эозинофилов. Именно они являются традиционными партнерами тучных клеток и базофилов в развитии аллергии, а характер воспаления недаром называют эозинофильным. Однако эозинофилия, как тканевая, так и сывороточная, сопровождает не только аллергические заболевания, потому ее дифференциальная диагностика является чрезвычайно актуальной.
Эозинофилию определяют, если количество эозинофильных гранулоцитов в периферической крови превышает 0,35 × 109/л. При этом нужно учитывать то, что эозинофилия крови не всегда коррелирует с тканевой. Выделяют реактивную эозинофилию, сопровождающую ряд болезней и патологических процессов, и клональную эозинофилию, предопределенную злокачественной пролиферацией кроветворных клеток. Длительная значительная эозинофилия вредно влияет на организм независимо от ее генеза [7].
Реактивная эозинофилия. Аллергическими болезнями, которые могут сопровождаться эозинофилией, являются: эозинофильная пневмония, аллергический ринит, бронхиальная астма, аллергический бронхолегочный аспергиллез, экзогенный аллергический альвеолит, аллергические реакции на медикаменты. В возникновении реактивной эозинофилии большое значение имеет паразитарная инвазия: анкилостомоз, аскаридоз, токсокароз, трихинеллез, описторхоз, стронгилоидоз, филяриоз, лямблиоз, токсоплазмоз, а также грибковые и ретровирусные инфекции [4, 7, 15].
Простая эозинофильная пневмония (синдром Леффлера) характеризуется инфильтратом размером от нескольких миллиметров до сантиметров, расположенным в обоих легких, минимальными клиническими проявлениями со стороны дыхательных путей и гиперэозинофилией. Этиологическим фактором синдрома Леффлера наиболее часто выступают такие паразиты, как Ascaris lumbricoides, Strongyloides stercoralis, Ancylostoma, Necator, Uncinaria. Инфильтрат может мигрировать по легочным полям, а через несколько недель рассасывается, не оставляя изменений в легочной ткани. Эозинофилию предопределяют не только гельминты с тканевой локализацией паразитов и личинок, но и миграция глистов, которые не адаптированы к организму человека — Toxocara canis, Toxocara catis и другие гельминты. Общение ребенка с котами и собаками нередко заканчивается его инфицированием. Анализ кала и мокроты на наличие паразитарной инвазии является первым в алгоритме диагностики «беспричинной» эозинофилии у детей. Гиперэозинофилия в этих случаях сопровождается повышением уровня общего ІgЕ [7].
Высокий уровень эозинофилии наблюдается также при диффузных болезнях соединительной ткани (СКВ, ювенильный ревматоидный артрит, прогрессирующий системный склероз, ЮДМ) и системных васкулитах [7]. Первичные иммунодефицитные состояния у детей тоже могут сопровождаться эозинофилией — синдром Вискотта — Олдрича, селективный дефицит IgA, гипер-IgE-синдром. Эозинофилия может возникать как реакция на применение некоторых медикаментов, в частности антибиотиков, сульфаниламидов, витаминов, препаратов никотиновой кислоты.
Клональная эозинофилия наблюдается при гемопатиях и онкологических заболеваниях, гиперэозинофильном синдроме, мастоцитозе, лейкемии, лимфоме. Для острой эозинофильной лейкемии характерна инфильтрация костного мозга молодыми формами эозинофилов [7]. Бласты идентифицируются с помощью специфической реакции на пероксидазу, резистентную к цианидам.
Особого внимания заслуживает так называемый идиопатический гиперэозинофильный синдром (ГЭС). Он характеризуется длительным повышением эозинофилов в периферической крови и инфильтрацией этими клетками многих органов и тканей, что предопределяет клиническую картину мультиорганного поражения. Синдром является гетерогенным по различным причинам его возникновения и патогенетическим механизмам, однако современные знания не позволяют определить отдельные заболевания, которые сегодня объединены термином ГЭС. Диагноз последнего оправдан в случаях, когда содержание эозинофилов в периферической крови больного составляет 1,5 × 109/л в течение более 6 месяцев и ассоциируется с органной патологией. Характерные изменения состава крови: количество лейкоцитов редко превышает 25,0 × 109/л, процент эозинофилов — 30–70. В периферической крови обнаруживают зрелые эозинофилы и их прекурсоры. В эозинофилах наблюдаются дегенеративные морфологические изменения: уменьшение числа и размеров зернистости, гиперсегментация ядра и наличие вакуолей в цитоплазме [7]. В костном мозге процент эозинофилов и их прекурсоров составляет 30–60. Количество миелобластов не увеличено, хотя в периферической крови больных ГЭС наряду с гиперэозинофилией увеличено содержание гранулоцитов и моноцитов. Прежде чем остановиться на диагнозе ГЭС проводится вся возможная на сегодня дифференциальная диагностика для исключения известных причин гиперэозинофилии. Клиническая картина ГЭС определяется такими неспецифическими симптомами, как общее недомогание, малопродуктивный кашель, боли в мышцах, повышение температуры тела, крапивница, нарушение зрения, сердечно-сосудистой и нервной систем, желудочно-кишечного тракта. Прогностически неблагоприятным является поражение сердца, что может привести к инвалидности и смерти.
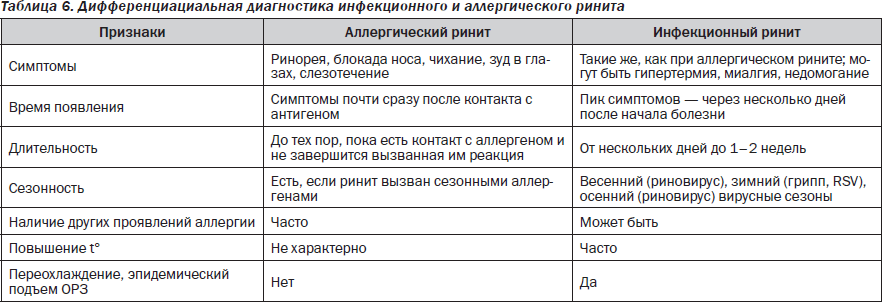
Инфекции. Чаще всего возникает необходимость дифференциации аллергических проявлений и детских инфекций, сопровождающихся экзантемами: кори, краснухи, скарлатины. Иногда инфекция Эпштейна — Барр проявляется сыпью, особенно если ребенок получает антибиотики пенициллинового ряда. Для всех детских инфекций характерна определенная стадийность высыпаний и их локализация, а также циклический характер течения инфекционного процесса. Кроме того, существуют и другие, свойственные каждой из них клинические симптомы и серологические маркеры, позволяющие в большинстве случаев поставить правильный диагноз в короткие сроки. Сложнее обстоит дело с дифференциальной диагностикой аллергического и инфекционного ринита, особенно в тех случаях, когда аллергическая риносинусопатия дебютирует первой из всех проявлений атопии. Более того, ребенок с аллергическим ринитом может инфицироваться и проявлять в клинической симптоматике смешанные признаки, свойственные обоим вариантам ринитов (табл. 6).
В этих случаях целесообразно назначать Н1-блокаторы рецепторов гистамина (антигистамины) ІІ поколения или их метаболиты, позволяющие влиять практически на все симптомы, в том числе и на заложенность носа, без использования или с минимальным использованием назальных деконгестантов (сосудосуживающих средств). При сохранении симптомов ринита необходимо провести аллергологическую диагностику, что позволит поставить окончательный диагноз.
В заключение следует отметить, что в практической деятельности детский аллерголог часто сталкивается с ситуациями, когда для установления истинного диагноза нужна широкая дифференциальная диагностика, которая базируется прежде всего на глубоких знаниях не только сугубо аллергической патологии, но и смежных состояний, что еще раз подчеркивает мультидисциплинарный характер аллергологии.
1. Дедишин Л.П. Системні васкуліти у практиці дитячого алерголога // Алергія у дитини. — 2007. — № 3. — С. 29-31.
2. Детская аллергология: Руководство для врачей / Под ред. А.А. Баранова и И.И. Балаболкина. — М.: Издательская группа «ГЭОТАР-Медиа», 2006. — 688 с.
3. Детская ревматология: Руководство для врачей / Под. ред. А.А. Баранова и Л.К. Баженовой. — М.: Медицина, 2002. — 335 с.
4. Жерносек В.Ф., Дюбкова Т.П. Аллергические заболевания у детей: Руководство для врачей. — Минск: ООО «Новое знание», 2003. — 335 с.
5. Иммунодефицитные состояния / Под ред. проф. В.С. Смирнова и проф. И.С. Фрейдлин. — СПб.: Фолиант, 2000. — С. 91-118.
6. Клинические рекомендации. Аллергология-2006. — М.: Издательская группа «ГЭОТАР-Медиа», 2006. — 227 с.
7. Ласиця О.Л., Ласиця Т.С., Недельська С.М. Алергологія дитячого віку: навчально-методичний посібник. — К.: Книга плюс, 2004. — 367 с.
8. Лыскина Г.А. Ювенильные формы системных васкулитов // Здоров’я України. — 2003. — № 22 (33). — С. 40.
9. Лыскина Г.А., Рябова Т.В., Маслиева Р.И. Ювенильный дерматомиозит: клиника, диагностика, течение // Педиатрия. — 2003. — № 3. — С. 77-83.
10. Марушко Т.В. Особенности диагностики и лечения неспецифического аортоартериита у детей // Здоровье ребенка. — 2007. — №6 (9). — С. 57-61.
11. Нагуа С.М., Гершвин М.Э. Секреты аллергологии и иммунологии: Пер. с англ. / Под ред. акад. РАМН Р.М. Хаитова. — М.: БИНОМ, 2004. — 319 с.
12. Наместникова О.Г., Кривошеев О.Г. Синдром Черга — Страусс: клиника, диагностика, прогноз, лечение // Medicus amicus. — 2007. — № 1. — С. 16-17.
13. Педиатрия 2005–2006: Клинические рекомендации / Под. ред. акад. РАМН А.А. Баранова. — М.: ГЭОТАР-Медиа. — 2005. — 272 с.
14. Прохоров Е.В., Челпан Ю.А., Сорока Ю.А. Диагностические критерии и особенности течения синдрома Чарга–Стросса у детей // Современная педиатрия. — 2004. — № 4 (5). — С. 170-171.
15. Пыцкий В.И., Адрианова Н.В., Артомасова А.В. Аллергические заболевания: Монография / Под ред. В.И. Пыцкого. — 3-е изд., перераб. и дополн. — М.: Триада-Х, 1999. — С. 343-352.
16. Семенкова Е.Н. Системные некротизирующие васкулиты // Русский врач. — 2001. — № 2. — С. 54-65.
17. Стефани Д.В., Вельтищев Ю.Е. Иммунология и иммунопатология детского возраста: Руководство для врачей. — М.: Медицина, 1996. — 384 с.
18. Ткаченко С.К. Алергічні маски ревматичних хвороб / Алергологія дитячого віку: проблеми і перспективи. — Матеріали обласної науково-практичної конференції, присвяченої 5-й річниці Львівського міського дитячого алергологічного центру. — Львів, 2005. — С. 14-19.
19. Чоп’як В.В., Ліщук-Якимович Х.О., Потьомкіна Г.О., Синенький О.В. Підходи до діагностики синдрому Чардж — Стросса: власні спостереження // Therapia. 2007. — № 3. — C. 67-69.
20. Guillevin L., Pagnoux C., Mouthon L. Churg-Strauss syndrome // Semin. Respir. Crit. Care Med. — 2004. — № 25 (5). — Р. 535-545.
21. Sable-Fourtasson R., Cohen P., Mahr A., Pagnoux C., Mouthon L., Jayne D. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome // Ann. Intern. Med. — 2005. — № 143 (9). — P. 632-638.
22. Solans R., Bosch J., Perez-Bocanegra C., Selva A., Huguet P. Churg-Strauss syndrome: outcome and long-term follow-up of 32 patients // Rheumatology. — 2001. — № 40. — P. 463-471.