
Газета «Новости медицины и фармации» 15(335) 2010
Вернуться к номеру
Фосфорно-кальциевый обмен при ХБП
Авторы: Д.Д. Иванов, д.м.н., профессор, НМАПО им. П.Л. Шупика
Версия для печати

Нарушения фосфорнокальциевого обмена, развивающиеся в результате ХБП, классифицируются KDIGO как костноминеральные нарушения — mineral bone disorder CKDMBD (рис. 1).

Нарушения фосфорнокальциевого обмена являются одной из основных составляющих прогрессирующего поражения почек, формируя два основных ведущих синдрома: резорбцию костной ткани и эктопическую кальцификацию. Наиболее неблагоприятной является кальцификация сердечнососудистой системы, клиническая значимость которой состоит в фатальном повышении рисков кардиоваскулярной смертности (рис. 2).
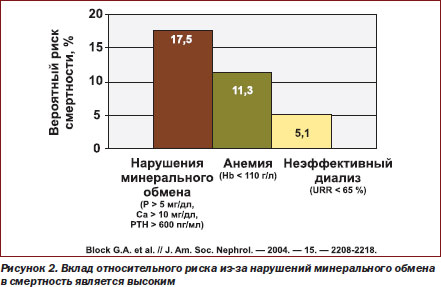
Костноминеральные нарушения ХБП несут в себе независимые факторы риска сердечнососудистой смертности: гиперфосфатемию, гиперкальциемию, повышение паратиреоидного гормона.
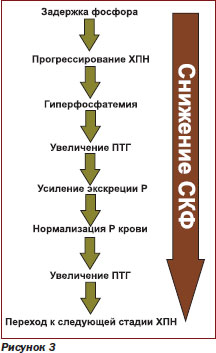
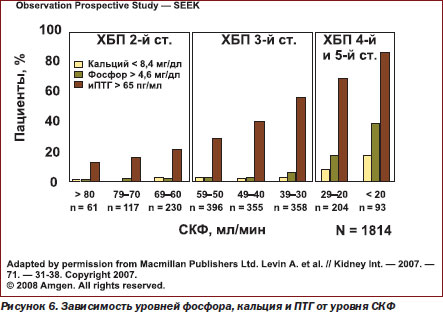
Началом развития костноминеральных нарушений является снижение способности почек к экскреции фосфатов на фоне практического неизменного их всасывания в кишечнике. Снижение СКФ (< 60 мл/мин) и секреторной функции канальцев приводит к совокупному уменьшению выведения фосфатов. Гиперфосфатемия является мощным фактором увеличения продукции паратиреоидного гормона (ПТГ), который компенсаторно усиливает экскрецию фосфатов, одновременно стимулируя выход кальция из костей и увеличение синтеза витамина D. Однако гиперкальциемии не наступает, чему способствует контроль со стороны тиреокальцитонина, который также усиливает и секрецию фосфора. Таким образом, при ХБП 2– 3й ст. уровень фосфора и кальция остается нормальным, а паратиреоидного гормона постепенно повышается (рис. 3).
По мере прогрессирования почечной недостаточности развивается резистентность канальцевого аппарата почек к действию ПТГ. Для усиления экскреции фосфора организмом начинает более активно использоваться витамин D, увеличивающий выведение фосфатов, но, помимо этого, стимулирующий всасывание кальция (и в меньшей мере фосфора) в кишечнике и реабсорбцию кальция в почках. Напомним, что витамин D2 (эргокальциферол) поступает с пищей, витамин D3 (холекальциферол) образуется в коже под воздействием ультрафиолетовых лучей. Витамины D2 и D3 подвергаются первому гидроксилированию в печени, где в последующем и сохраняются его основные запасы. При этом D3 превращается в 25гидроксихолекальциферол (25OHD3) — кальцидиол, а D2 — в 25гидроксиэргокальциферол (25OHD2). Это превращение катализирует фермент 25гидроксилаза. 25гидроксикальциферолы — основная транспортная форма витамина D в организме. В плазме крови они (как и другие формы витамина) переносятся специфическим транспортным белком — транскальциферином. Второе гидроксилирование происходит в почках, где из 25OHD3 с помощью 1aгидроксилазы образуется биологически активный витамин D3 (1,25дигидроксихолекальциферол, или кальцитриол), а посредством 24гидроксилазы — 24,25(OH)2D3. Аналогично из 25OHD2 образуются 1,25(OH)2D2 и 24,25(OH)2D2. Наиболее активная форма D3, ответственная за его функции, — 1,25(OH)2D3. Его биологическое действие в 10 раз превышает активность других форм, поэтому, говоря о дефиците витамина D при ХБП, прежде всего имеют в виду недостаточный синтез активного метаболита витамина D3.
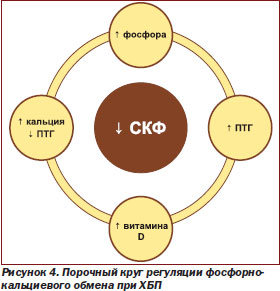
Важнейшие регуляторы, активирующие синтез 1,25(OH)2D3, — паратиреоидный гормон, эстрогены, пролактин, соматотропин и инсулин. Увеличение концентрации фосфора усиливает секрецию паратиреоидного гормона, который активирует в почках 1aгидроксилазу, в результате чего ускоряется синтез 1,25(OH)2D3 (рис. 4). Избыточное поступление Са2+ и Р с пищей подавляет синтез 1,25(OH)2D3, так как при этом его предшественник 25(OH)D3 превращается в почках в 24,25(OH)2D3, который стимулирует всасывание кальция и фосфора в кишечнике так же эффективно, как 1,25(OH)2D3, и одновременно стимулирует остеогенез и минерализацию костной ткани.
У пациентов с ХБП 3–4й ст. в различные сроки развивается сначала функциональный, а затем абсолютный дефицит витамина D3 (кальцидиола и затем кальцитриола) за счет уменьшения массы функционирующей паренхимы, продуцирующей 1aгидроксилазу. Основное патоморфологическое следствие развивающейся недостаточности витамина D — нарушение минерализации костной ткани. Биологическая активность витамина D измеряется в международных (интернациональных) единицах (ME); 1 ME соответствует активности 0,025 мкг эрго или холекальциферола. Содержание D2 и D3 в продуктах питания невелико. Например, в печени быка и сливочном масле соответственно 0,4 и 0,4– 3,2 МЕ/г. Исключение составляют жир печени трески и тунца, в которых этих витаминов содержится соответственно 50–350 и 40 000–60 000 МЕ/мл. Потребность человека в витамине D, составляющая 400 ME (10 мкг) в сутки, при достаточной и регулярной инсоляции обеспечивается фотохимическим синтезом D3 в коже или за счет поступления с пищей. Однако за счет пищевого рациона и воздействия ультрафиолетовых лучей компенсировать недостаток витамина у пациентов с ХБП невозможно. Поэтому в качестве лечебной опции пациентам с ХБП 3–4й ст. (KDOQI, 2003) рекомендуют целевое сывороточное содержание гидроксихолекальциферола (25(OH)D), то есть кальцидиола (неактивного витамина D3), в дозе > 75 ммоль/л, как и для всего населения. Его восполнение возможно за счет любых форм витамина D.
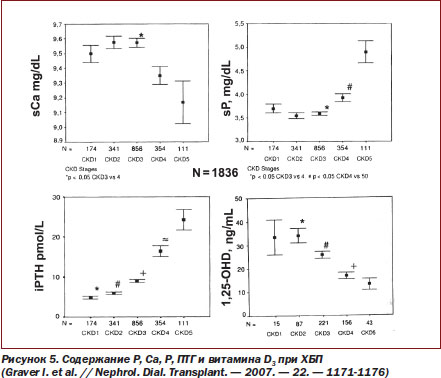
Таким образом, при ХБП 3–4й ст. уровень фосфора в крови остается еще нормальным, имея тенденцию к повышению, уровень кальция остается нормальным, имея тенденцию к понижению, уровень ПТГ постепенно повышается, уровни витамина D3 и его активного метаболита постепенно снижаются (рис. 5–6).
Следует отметить, что за счет взаимодействия с различными доменами в тканях организма витамин D3 реализует и другие эффекты, не связанные напрямую с синтезом ПТГ: снижение активности РААС и протеинурии, уменьшение левожелудочковой гипертрофии, защиту от атеросклероза и опухолей.
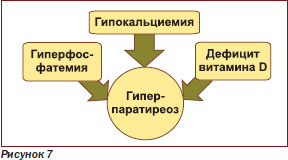
При достижении ХБП 5й ст. функциональный дефицит витамина D переходит в органический, так как изза резкого падения выработки 1aгидроксилазы практически прекращается синтез активного метаболита витамина — кальцитриола. В связи с этим для лечения ХБП 5й ст. (KDOQI, 2003) не рекомендуют назначать нативный витамин D, отдавая предпочтение активным формам витаминов D3 или D2. Формирующийся дефицит витамина D обусловливает развитие гипокальциемии. Таким образом, при ХБП имеют место нарастающая гиперфосфатемия, гипокальциемия, дефицит витамина D (особенно его активного метаболита) и все нарастающая выработка ПТГ (рис. 7).
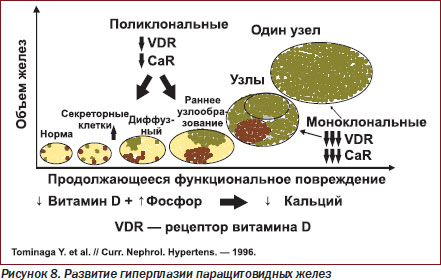
Гипокальциемия при дефиците витамина D способствует не только гиперпродукции ПТГ (вторичный гиперпаратиреоз, или гиперпаратиреоидизм), но и последующей гиперплазии паращитовидных желез и развитию их аденомы (рис. 8). Эта гиперплазированная ткань уже малочувствительна к регулирующему влиянию кальция и витамина D. Считается, что количество кальциевых рецепторов в узлах паращитовидной железы уменьшается почти на 60 %.
Кальций и витамин D3 практически независимо друг от друга регулируют выработку ПТГ через различные рецепторы: кальцийраспознающий рецептор (КР) и трансмембранный витаминD3рецептор (ВР). КР является поверхностным мембранным рецептором, находящимся в неактивном состоянии при низком содержании внеклеточного кальция, что определяет постоянный синтез ПТГ и является сигналом для увеличения количества КР. При повышении содержания кальция активируется КР, что быстро вызывает снижение количества ПТГ за счет угнетения его синтеза и секреции. Одновременно уменьшается экспрессия КР. Однако если низкое содержание кальция сохраняется во внеклеточной жидкости более нескольких минут, активизируются механизмы транскрипции и начинается пролиферация паратиреоидных клеток. Витамин действует значительно медленнее, так как для реализации эффекта ему необходимо проникнуть внутрь клетки паращитовидной железы, связаться с ядерным рецептором и только затем вызвать эффект подавления генной транскрипции, приводящей к снижению синтеза ПТГ. Таким образом, этиологическими факторами вторичного гиперпаратиреоза являются последовательно развивающиеся гиперфосфатемия, затем — дефицит активного витамина D3 и гипокальциемия.
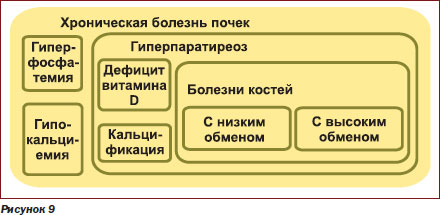
Весь каскад нарушений фосфорнокальциевого обмена, как было отмечено выше, формирует два жизненно неблагоприятных синдрома: эктопической кальцификации мягких тканей и сосудов и болезни костей (рис. 9).
Фосфор влияет на процессы прямой и непрямой кальцификации. Прямая кальцификация происходит за счет Ca ´ P продукта, а именно: повышенный внутриклеточный кальций образует ядро апатитного депозита в соединении с фосфором. Непрямая кальцификация происходит посредством развития гиперпаратиреоидизма: длительное увеличение уровня фосфора напрямую стимулирует синтез ПТГ и ведет к гиперплазии паратиреоидных желез, а ПТГ увеличивает базальный уровень внутриклеточного кальция.
Кальцификация фосфатами кальция начинается с интимы сосудов и в последующем захватывает мышечный слой, формируя жесткость сосудов (рис. 10). Кальцификация, происходящая в результате нарушений фосфорнокальциевого обмена, несколько отличается от поражения сосудов при кардиоваскулярных заболеваниях. Однако при ХБП имеют место оба типа нарушений.

В сердечной мышце также образуются очаги эктопических кальцификатов, что снижает нагнетательную способность миокарда. Процесс формирования эктопических кальцификатов стимулируется появлением остеобластподобных клеток, депозицией кристаллов фосфата кальция, увеличением FGF 23, фетуина А, матричных протеинов Gla и рядом других сигнальных молекул. Клиническими следствиями эктопической кальцификации сердечнососудистой системы являются развитие аритмий (внезапная смерть), сердечной недостаточности и повышение смертности от всех кардиоваскулярных событий (рис. 11). При этом следует помнить, что почечнозаместительная терапия не позволяет полностью исключить негативного влияния ограниченной функции почек на сердечнососудистую смертность.
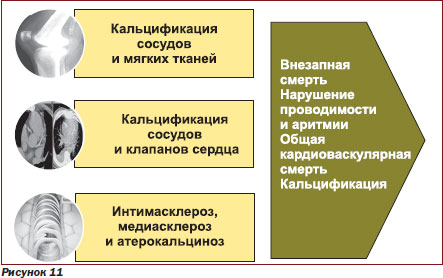
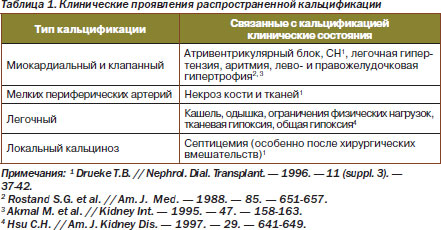
Среди других проявлений эктопической кальцификации наиболее часто документируют поражение легких, кожи и околосуставного аппарата (табл. 1).
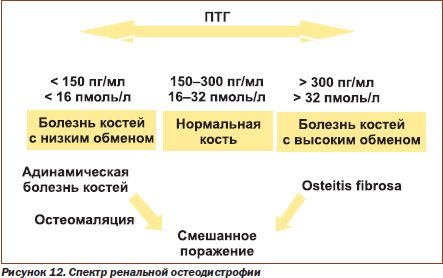
Второй жизненно важный патологический синдром — нарушение целостности костной ткани с формированием ренальной остеодистрофии (рис. 12).
Новая классификация (KDIGO, 2008) предполагает 3 ключевых процесса в оценке ренальной остеодистрофии: TMV (Turnover, Mineralization, Volume) (рис. 13–14).
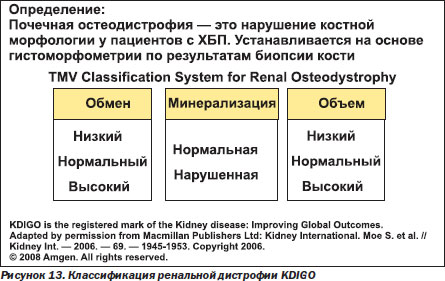

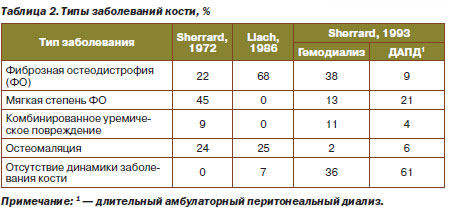
В зависимости от метаболизма костной ткани различают фиброзный остеит, остеомаляцию, адинамическую болезнь почек и смешанные нарушения (табл. 2). Решающей в установлении типа нарушений является биопсия кости с морфометрическим исследованием.

Руководство KDOQI (Guidelines on Bone Mineral Metabolism and Disease in CKD, 2003) рекомендует биопсию тазовой кости при следующих состояниях 5й стадии ХБП:
— переломы при минимальном воздействии или без видимой причины (патологические переломы);
— иПТГ в пределах 100–500 пг/мл при необъяснимой гиперкальциемии или выраженной боли в костях;
— предполагаемая алюминиевая болезнь костей, диагностированная на основании клинических симптомов или анамнеза использования алюминия.
Информативность рентгенологической двухфотонной абсорбциометрии (ДЕХА) у пациентов с ХБП окончательно не установлена и не несет специфической информации о метаболизме кости. DЕXA не коррелирует с гистологией костной ткани и частотой переломов. Поэтому DЕXA следует рассматривать с комплексе с другими биомаркерами кости и минерального обмена, клинической картиной и результатами гистологического исследования. В настоящее время для оценки состояния кости используется мультиспиральная томография, позволяющая получить трехмерное изображение вне влияния окружающих структур и распознать как кортикальные, так и трабекулярные структуры кости.
В практике упрощенно выделяют два вида нарушений: с высоким (фиброзный остеит) и низким обменом (адинамические нарушения, остеомаляция). В основе этих нарушений лежат различная регулирующая концентрация ПТГ, активность остеокластов/остеобластов и костной щелочной фосфатазы.
Высокие уровни ПТГ стимулируют остеобласты, обеспечивая высокий уровень обмена в костной ткани и формируя фибрознокистозный остеит. Высокий уровень обмена в костях приводит к образованию неупорядоченного остеоида, фиброзу и образованию кист, в результате чего истончается кортикальная кость, снижается прочность кости и повышается риск переломов (KDOQI, 2003). Этот вид костноминеральных нарушений сейчас встречается реже ввиду активной сопроводительной терапии витамином D.
Адинамические нарушения и остеомаляция характеризуются снижением костного обмена или ремоделирования с уменьшенным числом остеокластов и остеобластов, а также подавлением остеобластной активности. При остеомаляции наблюдается накопление неминерализованного костного матрикса, то есть увеличение объема остеоида, что может вызываться дефицитом витамина D или накоплением алюминия. Адинамическая остеодистрофия характеризуется снижением объема кости и ее минерализации и может вызываться накоплением алюминия или избыточным подавлением секреции ПТГ при помощи кальцитриола.
Адинамические нарушения кости (болезнь адинамической кости, адинамическая остеодистрофия) характеризуются нарушением образования матрикса кости, минерализации кости и снижением резорбции кости на фоне низкого/нормального уровня ПТГ и низкого уровня костноспецифической щелочной фосфатазы. Нередко адинамические нарушения возникают при передозировке кальциевых фосфатных биндеров и витамина D. Однако это не является основной причиной нарушений. Пациенты, страдающие адинамическими нарушениями кости, выглядят старше своего возраста, чаще болеют сопутствующими заболеваниями, имеют диабет, более выраженные атеросклеротические изменения и МИАсиндром (мальнутриция, воспаление, атеросклероз), долго лечились диализом.
Для лечения костноминеральных нарушений ХБП и вторичного гиперпаратиреоза используют диету, фосфатбиндеры, витамин D, а у диализных пациентов — низкокальциевые растворы (1,25 ммоль/л) при гиперкальциемии, высококальциевые (> 1,4 ммоль/л) — при гипокальциемии, а также кальцимиметики (только при почечнозаместительной терапии) при неэффективности терапии аналогами витамина D и выраженном гиперпаратиреозе.
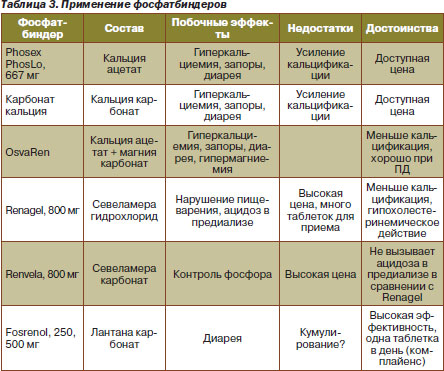
Начальным и необходимым этапом лечения является диета с суточным содержанием фосфатов 800–1000 мг. С учетом ряда социальных сложностей, в особенности у лиц молодого возраста, более простым методом является использование фосфатбиндеров — препаратов, молекулы которых, не всасываясь из кишечника, связывают фосфор и выводят его из организма в виде нерастворимых веществ (табл. 3). Алюминийсодержащие фосфатбиндеры, несмотря на их хорошую эффективность, в настоящее время не используются изза высокого риска кумуляции алюминия с последующим развитием энцефалопатии и поражения костей.
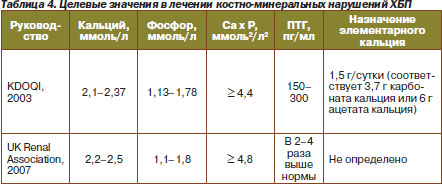
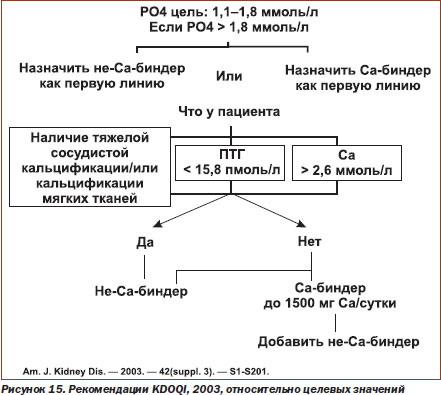
Современные руководства определяют целевые значения в лечении костноминеральных нарушений ХБП (табл. 4, рис. 15).
В настоящее время считается нецелесообразным использование соотношения Са х Р как некорректно характеризующего минеральные нарушения. Однако современные рекомендации KDOQI ждут своего представления в 2010 году.
Назначение активных метаболитов витамина рассмотрено в предыдущей лекции. Следует еще раз подчеркнуть, что терапевтическое окно 1,25(OH)2D3 достаточно узкое и лечение витамином D связано с повышенным риском кардиоваскулярных кальцификаций, которые частично могут быть связаны с гиперкальциемией, гиперфосфатемией, увеличением активности костной щелочной фосфатазы, остеопонтина и уменьшением секреции гладкомышечными сосудистыми клетками ПТГсвязанного пептида. Дозы 1,25(OH)2D3, оптимальные для восполнения дефицита гормона, но не развития токсических эффектов, все еще не установлены. Ежемесячный контроль ПТГ при использовании кальцитриола позволяет подобрать правильную дозу. Однако даже при передозировке токсический эффект лечебных доз 1,25(OH)2D3 потенциально обратим. Кроме того, токсический эффект лечебных доз 1,25(OH)2D3 может быть уменьшен при назначении кальцимиметиков.
Однако следует напомнить, что витамин D3, реализуя свой эффект, взаимодействует с целым каскадом сигнальных молекул, а именно: сывороточным витаминD3связывающим протеином, мембранным рецептором, 25гидроксивитаминD324гидкроксилазой (CYP24) и, наконец, ядерным рецептором. Современные препараты могут вызывать активацию рецептора витамина с помощью не только активного метаболита витамина D3, но и активного метаболита витамина D2 (1,25дигидроксивитаминD2), например парикальцитола (zemplar, abbott). При прямом сравнении с кальцитриолом гиперкальциемия и гиперфасфатемия остаются одинаковыми, но сроки сохранения гиперкальциемии несколько короче при назначении парикальцитола, что связывают с меньшей активацией КР в кишечнике. Признано целесообразным использование активных форм витамина D у пациентов, получающих диализ, и, возможно, после трансплантации почки.
Вместе с тем известно, что существует внепочечный синтез активного метаболита витамина D в макрофагах, эндотелии, предстательной железе, молочной железе и головном мозге. При снижении синтеза кальцитриола в почках названные ткани усиливают свой локальный синтез, хотя он малосравним с почечным. Локальная выработка 1,25(OH)2D3 модулирует ряд клеточных функций для продления жизни пациентов с ХБП (Holick M.F. // AJKD. — 2005). Поэтому целесообразность назначения обеих форм витамина, возможно, обусловлена не только заместительной функцией кальцитриола, но и обеспечением субстрата для независимых внепочечных эффектов, предупреждающих атеросклероз, гипертензию, иммунодефицит и развитие рака.
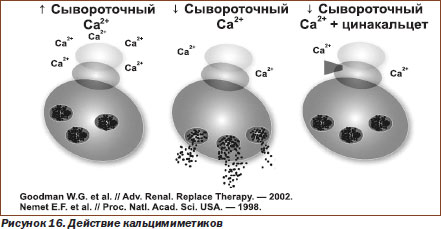
Кальцимиметики увеличивают чувствительность кальцийраспознающего рецептора и уменьшают уровень кальция и ПТГ (рис. 16, табл. 5). Различают кальцимиметики І (неорганические вещества, например Mg2+, Gd3+, аминогликозиды, спермин) и ІІ типов. Цинакальцет (mimpara 30, 60, 90 мг) — кальцимиметик ІІ типа, первый широко применяемый препарат у диализных пациентов с неэффективным результатом терапии витамином D. Суточная доза — 60–180 мг в один прием. При ХБП 3–4й ст. цинакальцет может вызывать гипокальциемию и гиперфосфатемию. Однако продолжается накопление клинического опыта применения цинакальцета у пациентов с ХБП 3–4й ст. и у посттрансплантационных больных. Кальцимиметики рекомендуются для длительного постоянного приема, при этом доза витамина D может быть уменьшена.
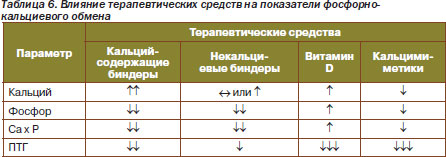
Таким образом, суммируя данные о влиянии терапевтических средств, можно сделать заключение, что коррекция фосфорнокальциевых нарушений требует должного искусства со стороны врача (табл. 6–7) (A.L.M. Francisco, F. Carrera).


Однако у ряда больных перечисленные мероприятия не приводят к нормализации/существенному уменьшению содержания ПТГ. Как правило, это обусловлено развитием гиперпластических узлов в паращитовидной железе, которые экспрессируют малое количество КР и ВР и поэтому не чувствительны к кальцию и терапии активным витамином D. В таких случаях рекомендуется визуализация паращитовидных желез. Для обнаружения и оценки размеров ПТГ используется УЗИ (+ цветной допплер), сцинтиграфия с Тс99m, МРТ и КТ. Если одна из желез > 1,0 см в диаметре по данным УЗИ или уровень ПТГ превышает 600–800 пг/мл, следует ожидать резистентности к терапии кальцитриолом. Увеличение в размерах более 3 желез с признаками узелковой гиперплазии требует рассмотрения вопроса о назначении цинакальцета или проведения паратиреоидэктомии оперативным путем или при помощи склерозирующей инъекции.
Полностью ознакомиться с материалами можно, приобретя книгу Д.Д. Иванова «Лекции по нефрологии». — Донецк: Издатель Заславский А.Ю., 2010
Список литературы находится в редакции